Loading the trait data
Starting with BEAST X, it is possible to import trait data directly without any sequence data.
To start, select the Create partition from trait... at the bottom of the panel.
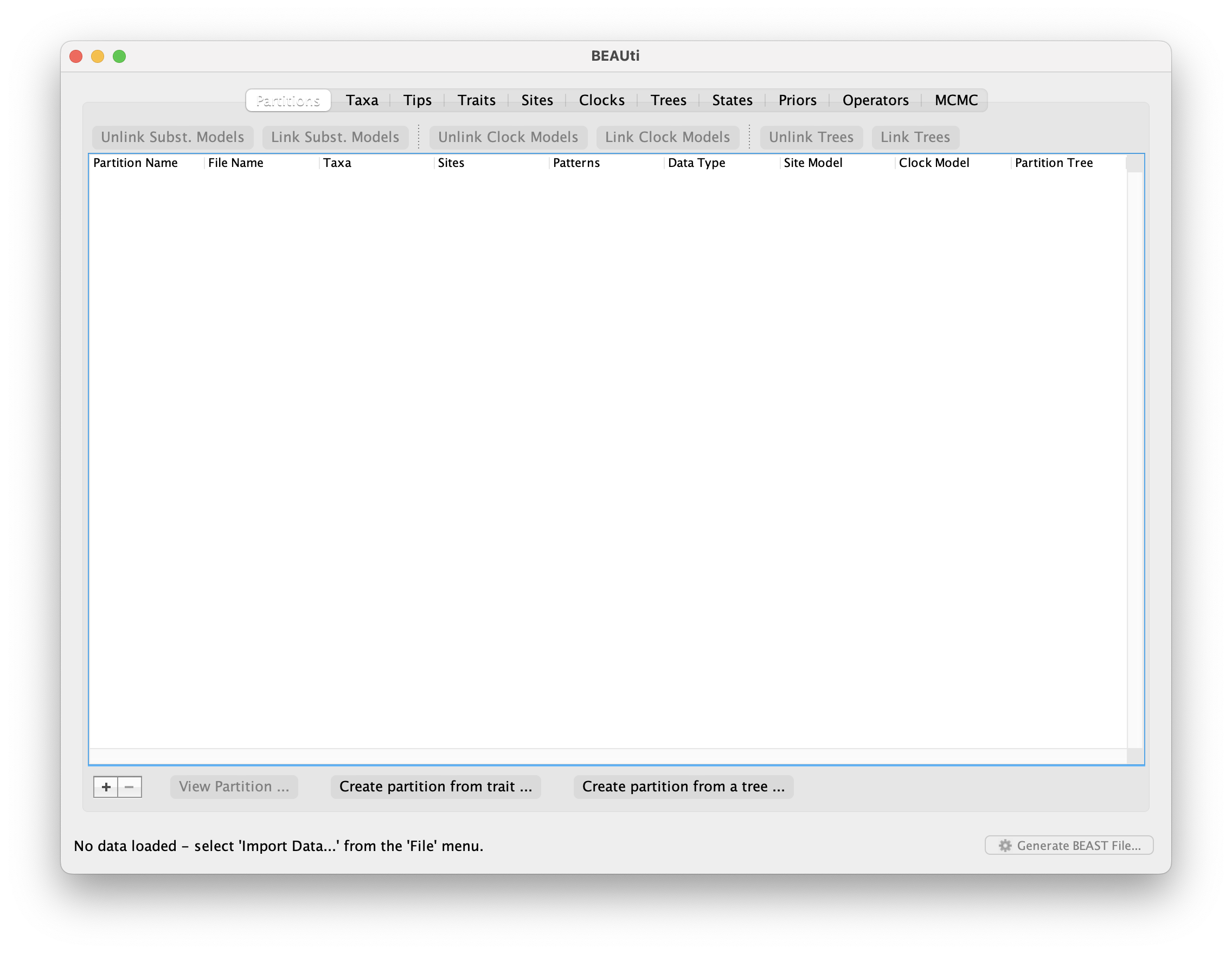
Next import the trait data from the file examples/continuousTraits/mammals.txt. This is a tab-delimited text file with the first column having taxon names (to match those in the sequence data) and each subsequent column having trait values. The first row contains the names of the traits (the first column of the first row is ignored and can be empty). Missing values can be coded as “NA” or “?”.
Highlight all the traits you want to include in the analysis (in this case all of them).
Select a name for the traits (in this case “life history”) and click OK.
If you want to treat traits independently, you can check the box New partition for each trait, but do not check that box for the purposes of this tutorial.
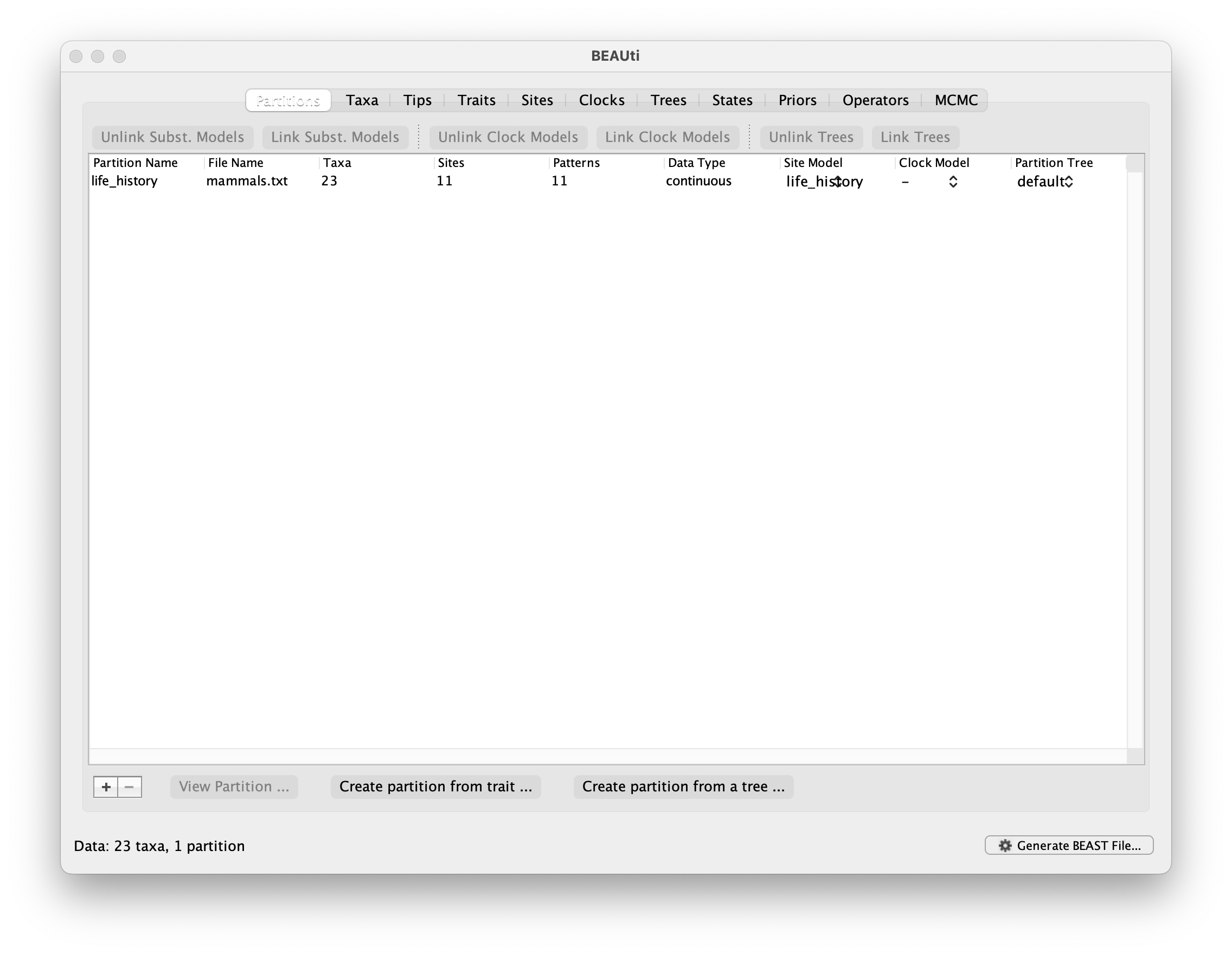
You should now see that the traits have been imported as a partition:
Setting up the trait model
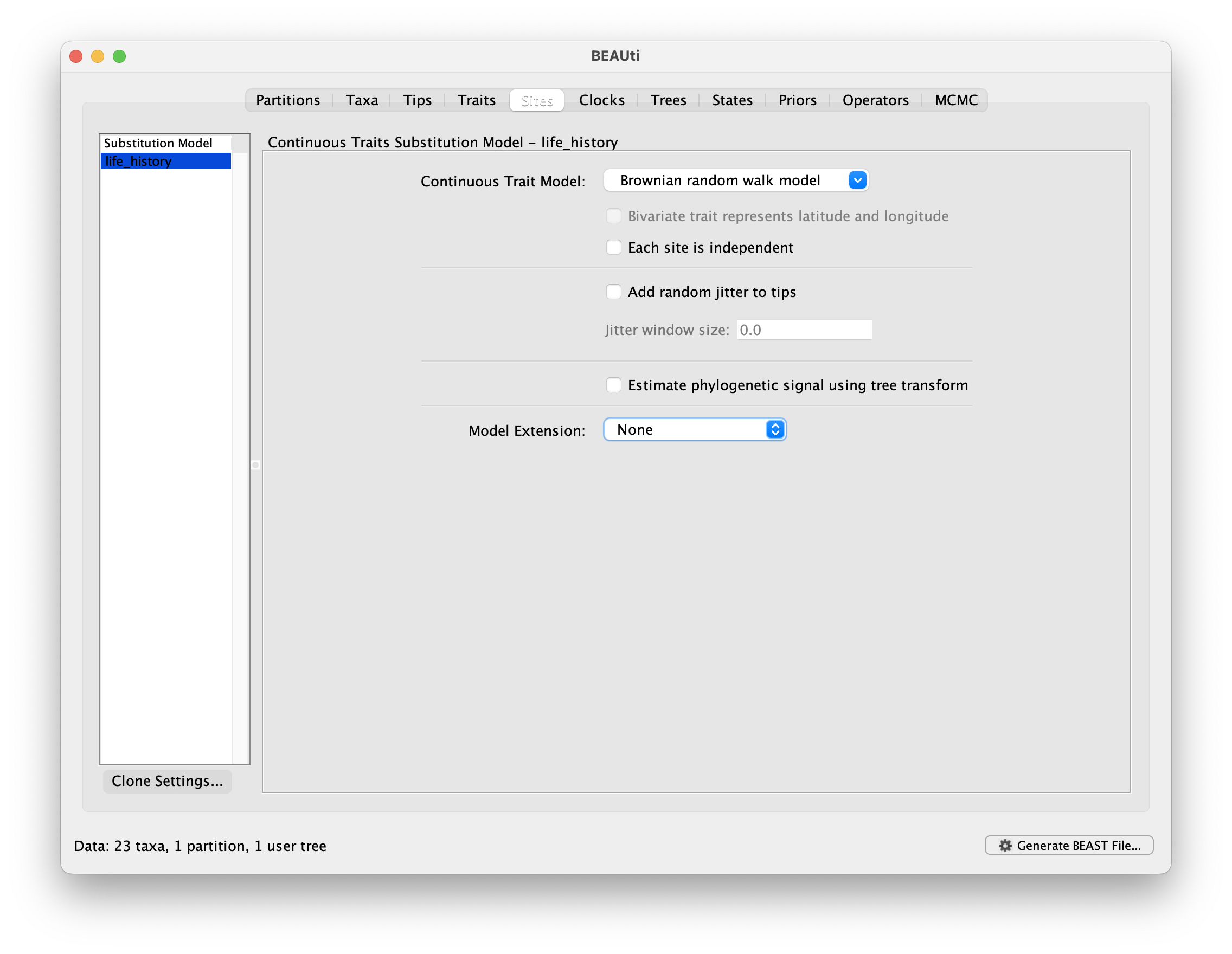
Switch to the Sites panel to select the evolutionary model for the trait partition.
This tutorial uses the default Brownian random walk model.
You can optionally specify a model extension.
The Residual variance extension assumes either 1) some measurement error in the observed traits or 2) that the process on the tree does not fully capture the variation in the traits.
This model permits estimates of trait-specific phylogenetic heritability.
The Latent factor model (also known as phylogenetic factor analysis / PFA) assumes a low-dimensional process on the tree that maps to high-dimensional traits.
For example, the mammals dataset has 11 traits, but it is possible that there are fewer (e.g., 2 or 3) evolutionary processes that are responsible for the variation in these 11 traits.
PFA maps the 11-dimensional traits to k-dimensional factors that evolve on the tree.
This model is particularly useful when computation is constrained by a large number of traits.
Specifically, non-PFA models scale cubically in the number of traits and may be computationally intractable with many traits.
However, PFA scales linearly and remains computationally feasable for even thousands of continuous traits.
We do not select either model extension for this tutorial, but want users to be aware of their existence.
Importing the tree
Often trait-only analyses rely on a fixed tree that has been previously inferred using sequence data or other features.
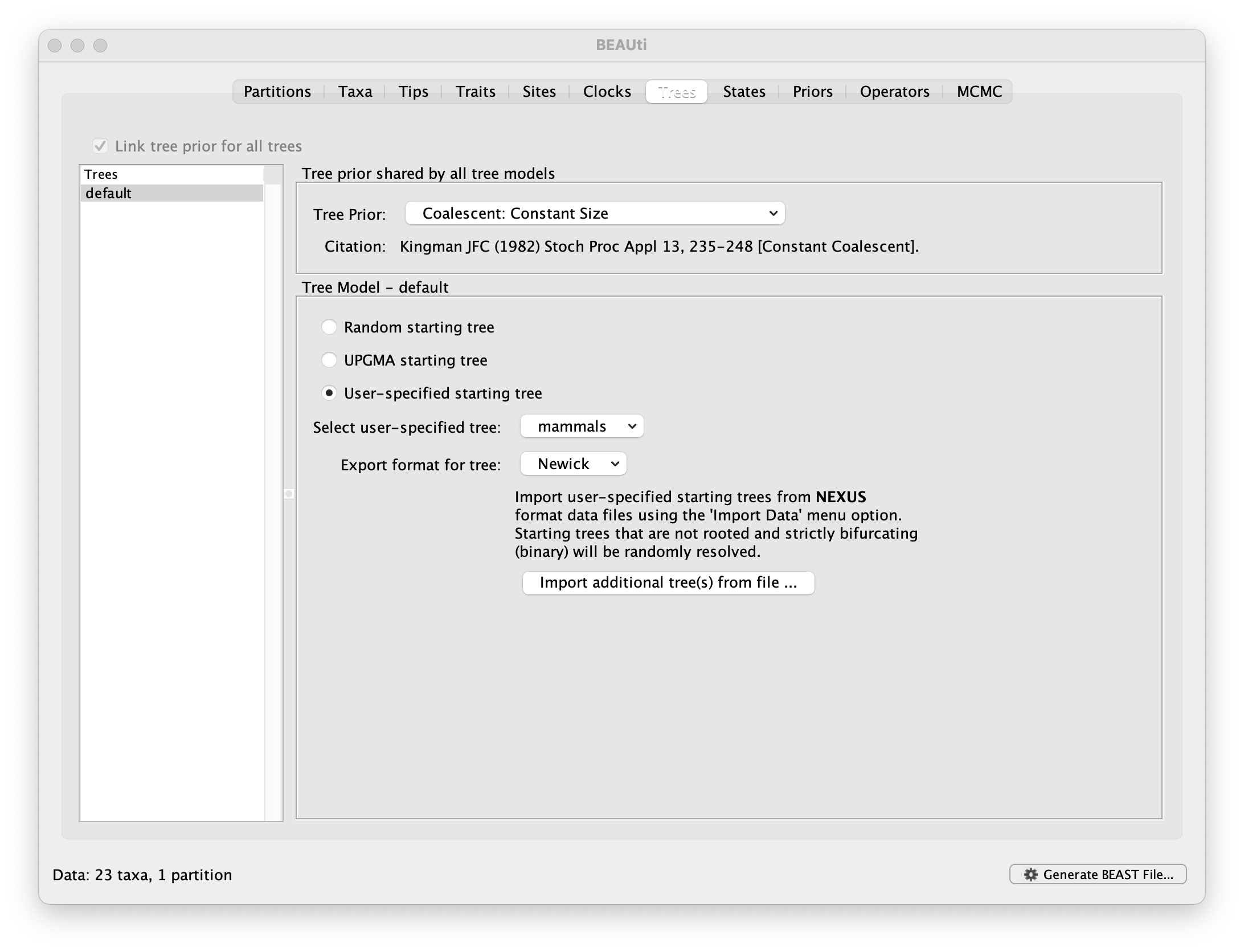
You can import a tree as a newick format (extension .nwk) from the Trees panel, by selecting Import additional tree(s) from file....
After importing the examples/continuousTraits/mammals.nwk file, you can set the Tree Model the User-specified starting tree: mammals
Disabling tree sampling
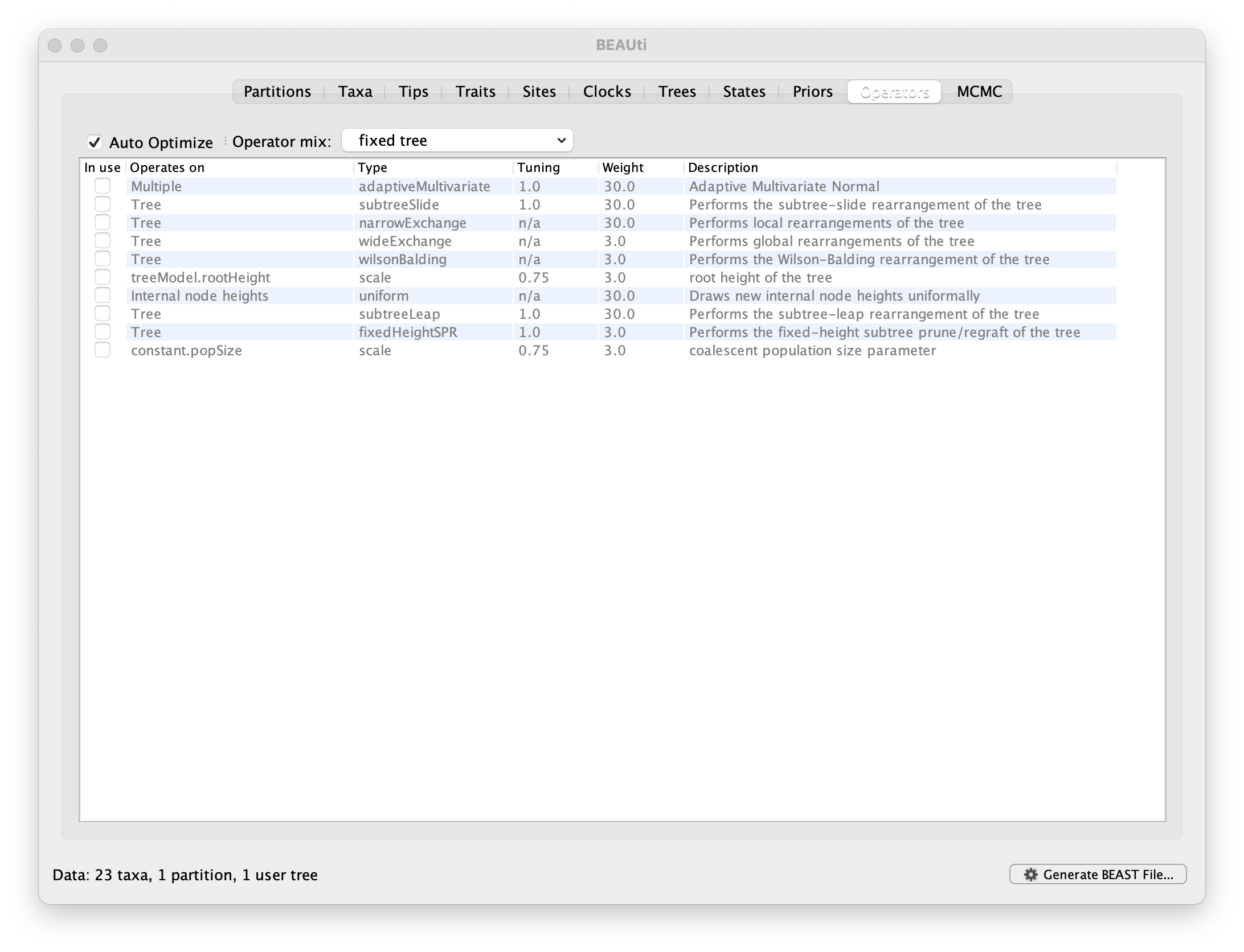
As mentioned above, many trait-only analyses rely on fixed trees.
To ensure that BEAST keeps the supplied tree fixed and does not sample from the posterior of trees, go to the Operators tab and select fixed tree in the Operator mix dropdown menu.
Generating the XML and Analysing the Data
See the Analysing Continuous Traits tutorial for information on generating the BEAST XML and analysing the data.
The process is the same, except that many of the tree-specific paramters (e.g. treeModel.rootHeight will be constant).