Taxon sets are groups of taxa or sequences that can be used in a number of ways within BEAST. For example, a taxon set can denote a clade in the tree (i.e., a common ancestor and all its descendants) that have a specific rate of evolution (i.e., a local clock model). It could specify a group of tips to which some model is applied. It can be used to specify a node in the tree — the TMRCA of that set of taxa — to which a calibration prior will be applied.
Taxon sets are defined in the Taxa panel of BEAUTi and can be created once some data has been imported. For example, import this file (IVA_PB1.xml - a set of 124 human, swine and avian influenza PB1 genes).
Creating a taxon set in BEAUti
Once loaded, switch to the Taxa panel, and then click the ‘plus’ at the bottom of the left hand table. A new taxon set will be created in this table, called ‘untitled1’:
Clicking on the cell with ‘untitled1’ will allow you to edit this name into something meaningful (in this case ‘human’).
The two tables on the right then define the taxa that are included and excluded from the set (to start with the set is empty so all the taxa are in the ‘excluded’ table). We want to create a taxon set of all the human viruses. We could select each in turn and move them into the included table but that would be tedious. Instead we can type ‘Human’ into the search box above the excluded table and all the taxon labels with the word ‘Human’ will be selected:
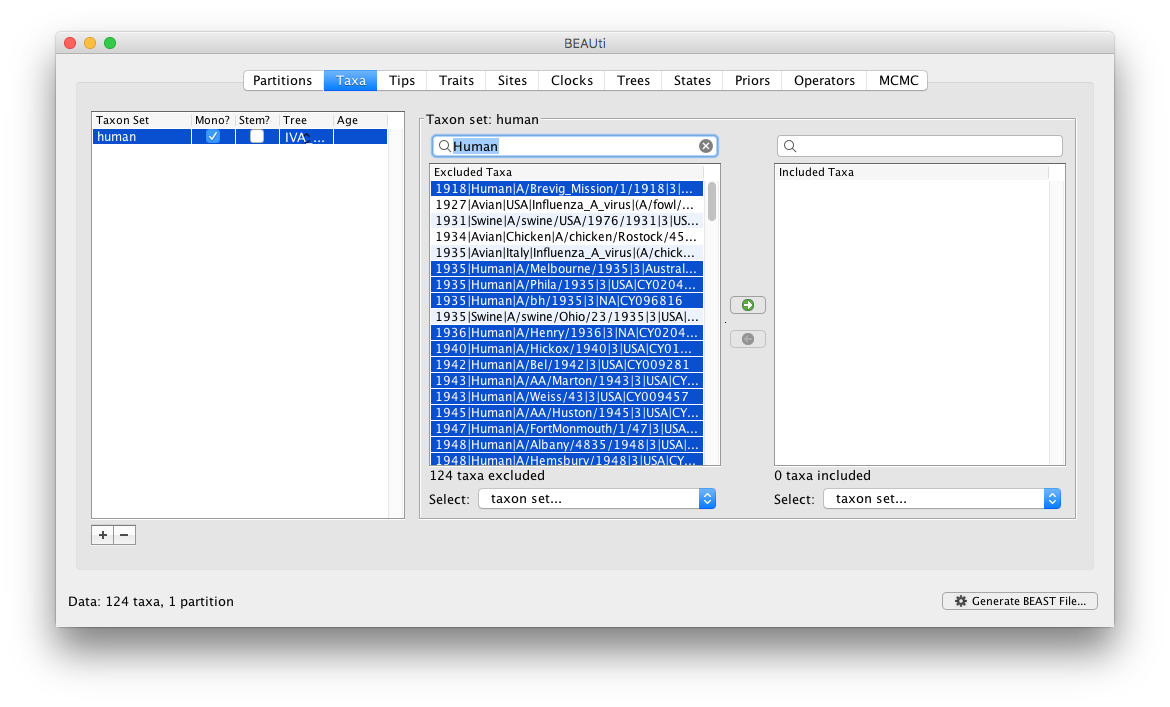
You can then just click the green arrow button in the middle to move these into the ‘included’ table:
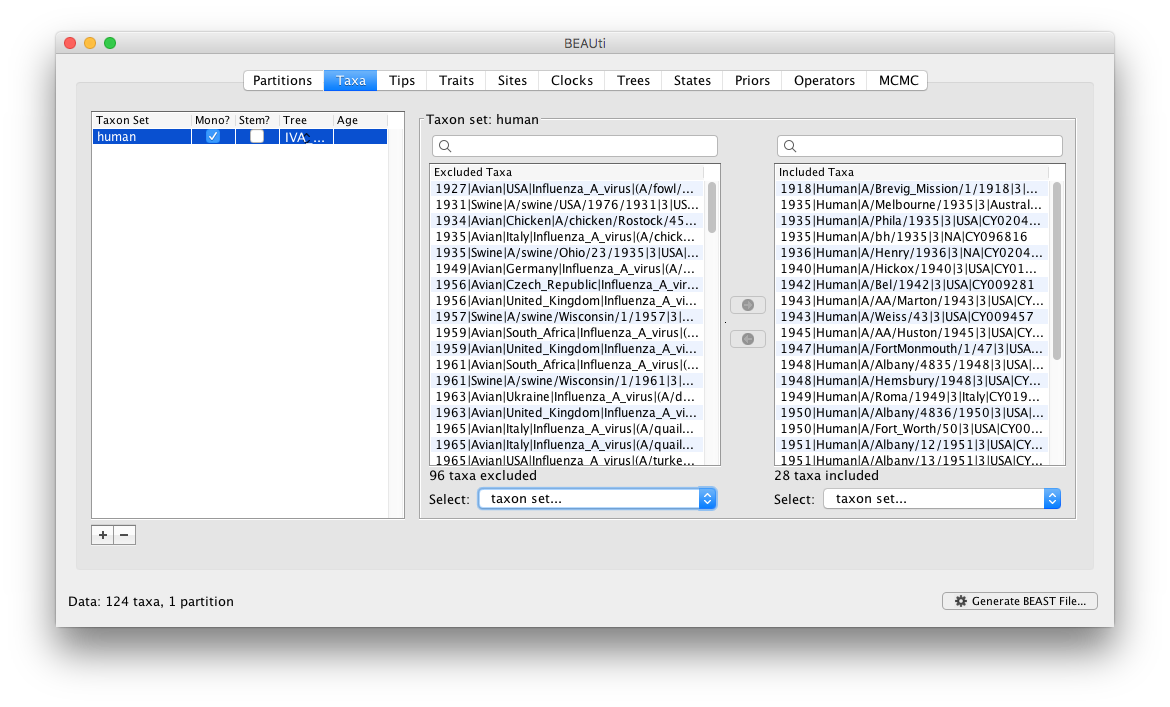
You can also do the same in the search box above the ‘included’ table — i.e., select all sequences from Ohio and remove them.
Importing a taxon set from file in BEAUti
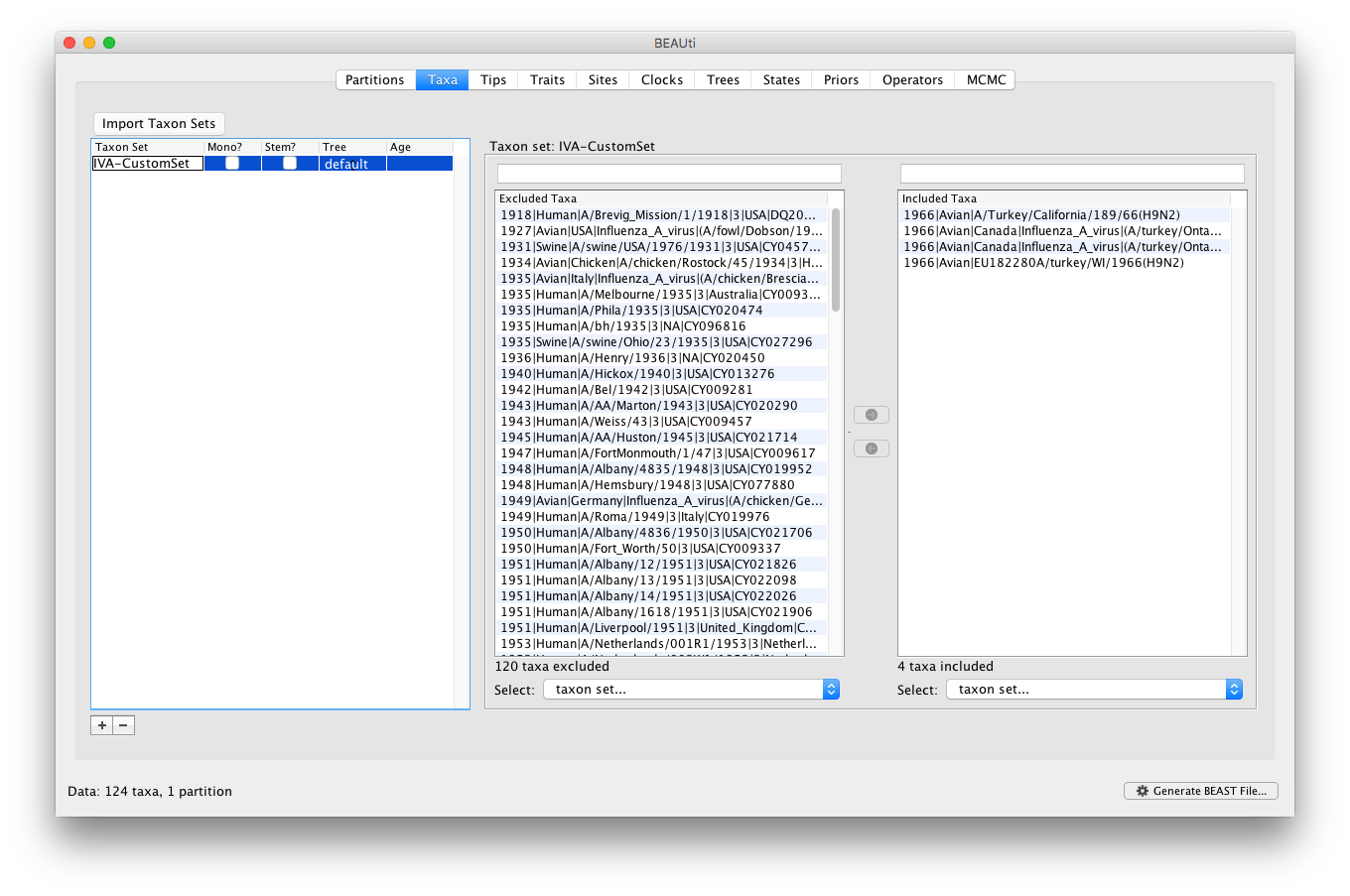
Suppose that no taxon sets have yet been defined or imported for this particular example. In order to import a taxon set from file into BEAUti, you can use a simple text file that contains the IDs (typically the sequence names) of the sequences you want to include in a specific taxon set.
For this example, this could be a .txt file that has the following 4 sequence names (i.e. they have to be the same IDs as in the main IVA_PB1.xml file):
1966|Avian|A/Turkey/California/189/66(H9N2)
1966|Avian|Canada|Influenza_A_virus|(A/turkey/Ontario/6213/1966(mixed))
1966|Avian|Canada|Influenza_A_virus|(A/turkey/Ontario/7732/1966(H5N9))
1966|Avian|EU182280A/turkey/WI/1966(H9N2)
If the file name for this is IVA-CustomSet.txt, then the newly imported taxon set will be named IVA-CustomSet in the Taxon Set panel:
Options in the Taxon Set table
The Select menus at the bottom of each of the ‘include’ and ‘exclude’ table allow you to select previously defined taxon set (in case you want to create the union of two sets or subtract one from the other).
Once the taxon set is defined, there are a few options in the Taxon Set table that you can specify.
- Mono?
- This specifies that you wish this taxon set to be forced to be monophyletic within your tree. This will mean that the random starting tree will have this set as a monophyletic clade and BEAST will never be allowed to break this constraint (by adding other taxa into the clade).
- Stem?
- This is used when the taxon set is being used to specify a subtree (i.e., a clade or a TMRCA). If click then this means the subtree includes the branch above the clade or TMRCA — the stem branch.
- Tree
- This specifies which tree the taxon set refers to.
- Age
- This specifies an initial age for the TMRCA of the taxon set in the starting tree. This makes it easier to ensure this node age lies within a calibration prior with hard bounds.