Some notes on pattern compression and speeding up BEAST analysis
Pattern compression for fun and profit
by Andrew Rambaut & Philippe Lemey
To calculate the likelihood of a tree given a sequence alignment, the log likelihood of each site (column in the alignment) is evaluated independently and then summed across all sites in the alignment. The likelihood of any sites that have exactly the same nucleotides for each taxon will be the identical (assuming they have been assigned the same substitution model and rate). So, in practice, BEAST ‘compresses’ the alignment into a set of unique patterns of nucleotides (referred to as just ‘patterns’) and the count of how many times they occur. Thus, the likelihood for the alignment is the sum of the likelihood for each pattern times their counts. Obviously, this trick provides considerable speed improvements over just naively calculating the likelihood for each site independently and is used by most (all?) phylogenetic software.
In the latest version (v10.5.0), BEAUti now shows the number of unique site patterns for a loaded alignment as well as the original length of the alignment:
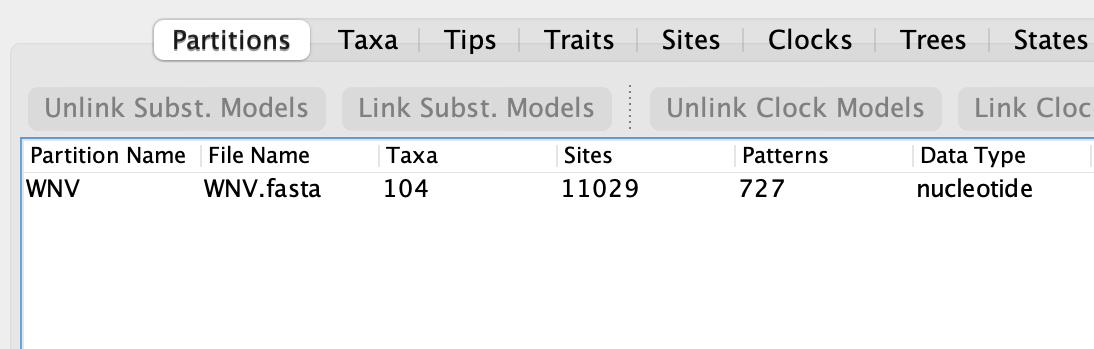
In this example using the file WNV.fasta from the examples/Data folder shows that this alignment of West Nile Virus genomes is compressed from over 11,000 nucleotides to only 727 unique site patterns - a compression of over 15 times with a commensurate improvement in likelihood calculation speed). The less variable the sequences in the alignment are, the greater the level of compression will be. This is because almost all the compression comes from the ‘constant’ sites that have the same nucleotide for every taxon. Other sites with nucleotide changes are unlikely to share exactly the same pattern with another such that they can be compressed.
Here is the top 15 most common site patterns for the WNV.fasta alignment:
| count | pattern |
|---|---|
| 2821 | GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 2635 | AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA |
| 2025 | TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT |
| 1963 | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC |
| 60 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANAAAAAAAAAAAAAAA |
| 56 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGNGGGGGGGGGGGGGGG |
| 49 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCNCCCCCCCCCCCCCCC |
| 35 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 30 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC |
| 26 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA |
| 24 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTNTTTTTTTTTTTTTTT |
| 21 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT |
| 11 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANNNNAAAAAAAAAAAAAAA |
| 11 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTNNNTTTTTTTTTTTTTTTT |
| 10 | NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCNNNNCCCCCCCCCCCCCCC |
The full table can be seen here. The first observation (as expected) is that the constant sites are the 4 most common patterns by a large margin (they make up 9,444 of the 11,029 sites in the alignment. The second is that most of the other patterns that are at high frequency are ones that are constant except for ambiguities (Ns) in some of the taxa. This is a feature of next generation sequencing where areas of low read coverage are filled in with Ns. Ambiguities are treated as unknown nucleotides in BEAST and are efficiently dealt with in the likelihood calculations done by BEAGLE.
This second observation offers an opportunity for further compression of the patterns. If we assume that patterns that are constant except for ambiguities are, in fact, constant then we can just include them in the counts for the respective constant patterns. For example, in the table above the 5th pattern comprises only As and Ns so might be considered as a constant A pattern. The 10th and 13th patterns are also constant As but differ in their pattern of Ns so would also be compressed similarly. Doing this for the WNV.fasta alignment goes from 727 unique site patterns down to 604 ‘ambiguous constant’ site patterns – a reduction of 17%. When run in BEAST with some standard models/settings the time drops from 11.2 minutes to 9.9 minutes for a reduction in run-time of 12%.
For other data sets, a much larger saving can be achieved. Here are a few examples:
| Virus | Dataset | Sequences | Sites | Unique patterns | Compressed ambiguous | Factor |
|---|---|---|---|---|---|---|
| SARS-CoV-2 | B.1.1.7 | 976 | 29409 | 2079 | 918 | 2.3x |
| SARS-CoV-2 | omicron_BA1 | 1000 | 29409 | 1528 | 485 | 3.2x |
| Ebolavirus | Makona_1610 | 1610 | 18992 | 7926 | 2267 | 3.5x |
| Mpox virus | cladei_reservoir | 60 | 196858 | 3701 | 386 | 9.6x |
In general, we would expect an increase in likelihood evaluation speed to improve by similar factors (with some allowance for overheads).
In BEAST X v10.5.0 a new command line option allows this feature to be switched on:
-pattern_compression ambiguous_constant
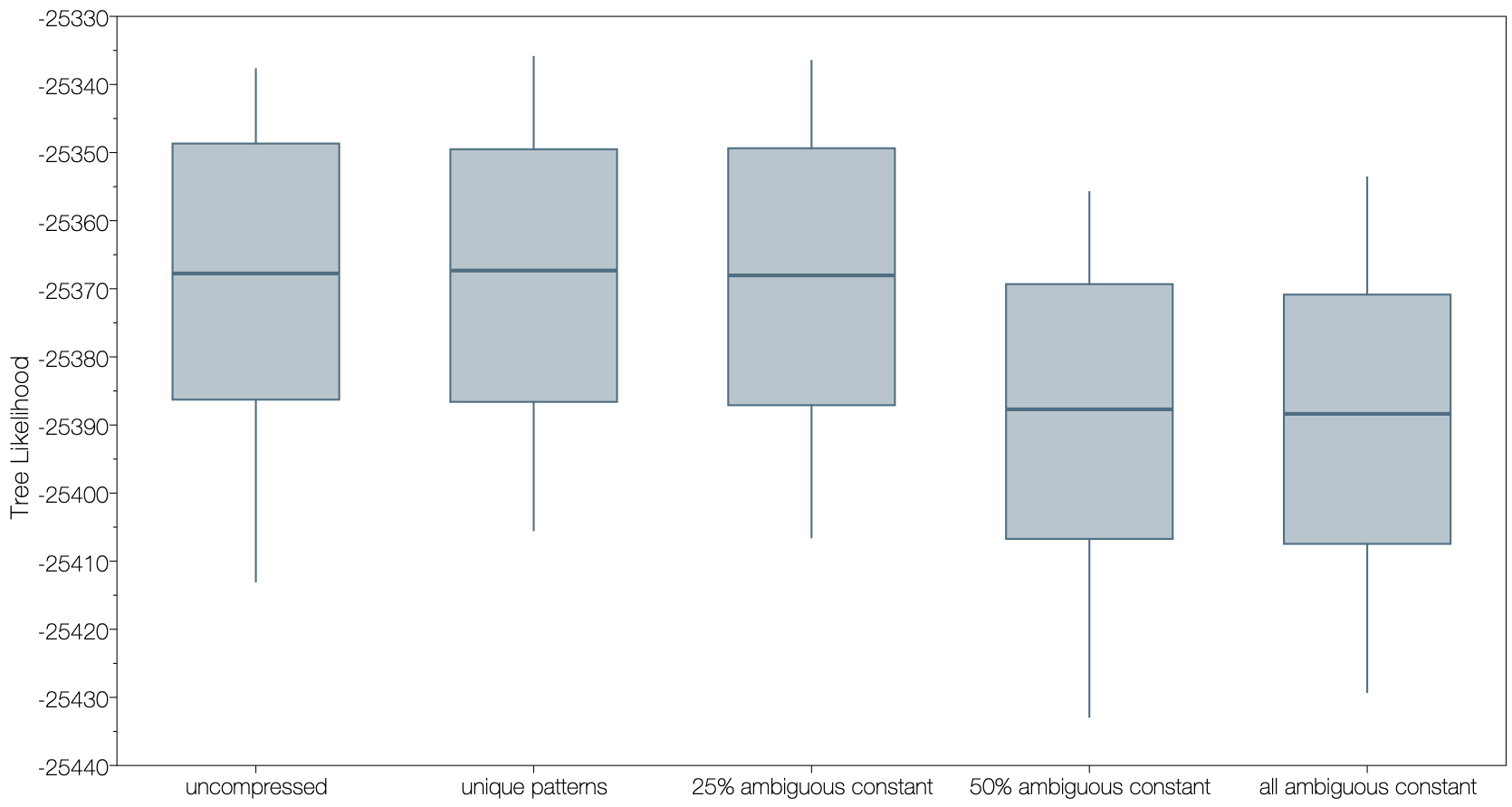
It is important to note that this feature is an approximation to the full likelihood calculation. In testing it appears to be a good approximation with only small differences in the estimated values of the parameters. A second option, -ambiguous_threshold, specifies the maximum proportion of nucleotides in the pattern that can be ambiguous and still be considered to be constant. This can help reduce the effect of the approximation whilst still allowing considerable savings in run-time. For example, compressing all constant sites irrespective of the proportion of ambiguous nucleotides produces a drop in the log likelihood of the tree (Figure 2, all ambiguous constant). Reducing the -ambiguous_threshold to 0.25 (which is the default value if not specified) returns a tree likelihood similar to the normal compression approach (Figure 2, unique patterns).
| Compression | Threshold | Pattern count | Run time |
|---|---|---|---|
-pattern_compression |
-ambiguous_threshold |
||
off |
n/a | 11,029 | |
unique |
0.0 | 727 | 11.2 minutes |
ambiguous_constant |
0.25 | 659 | 8.1 minutes |
ambiguous_constant |
0.5 | 608 | 7.8 minutes |
ambiguous_constant |
1.0 | 604 | 7.3 minutes |