Introduction
The first step is to convert an alignment file in FASTA format into a BEAST XML input file. This is done using the program BEAUti (which stands for Bayesian Evolutionary Analysis Utility). This is a user-friendly program for setting the evolutionary model and options for the MCMC analysis. The second step is to run BEAST using the input file that contains the data, model and settings. The final step is to explore the output of BEAST in order to diagnose potential problems and to summarise the results.
To undertake this tutorial, you will need to download three software packages in a format that is compatible with your computer system (all three are available for Mac OS X, Windows and Linux/UNIX operating systems):
Running BEAUti
The program BEAUti is a user-friendly program for setting the model parameters for BEAST. Run BEAUti by double clicking on its icon.
Loading the sequence data file
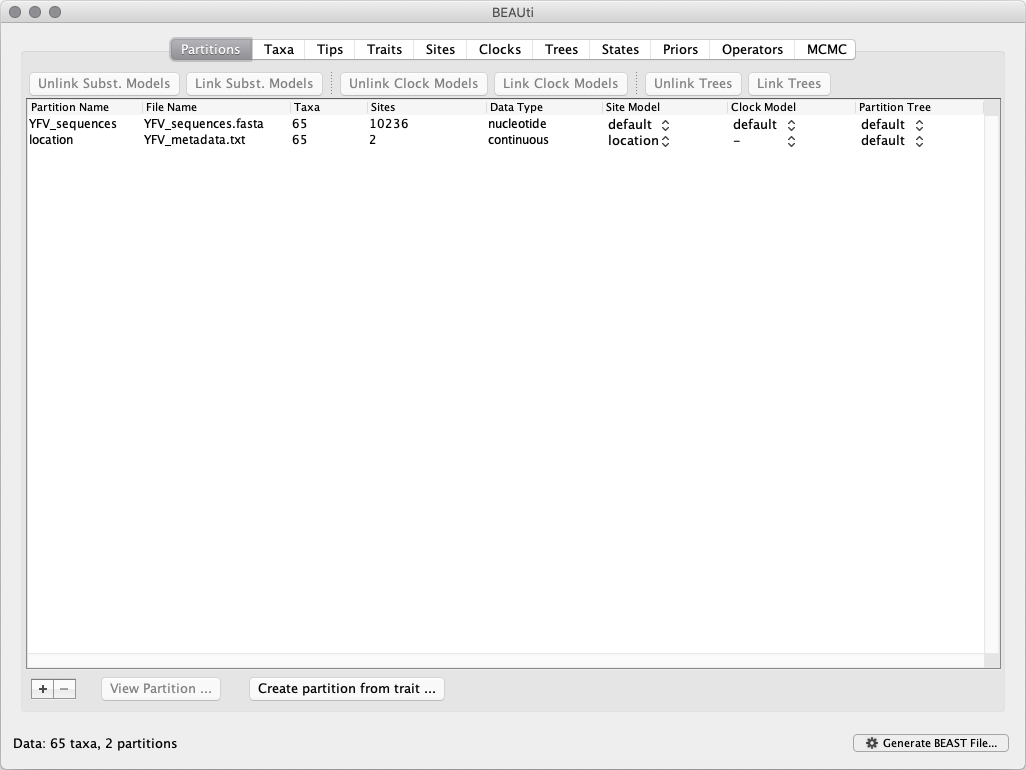
To load the alignment, simply select the Import Data... option from the File menu and browse to the ‘YFV_sequences.fasta’ file to load it. This file contains an alignment of 65 YFV genomes 10,236 nucleotides in length. Once loaded, the sequence data will be listed under Partitions as shown in the figure:
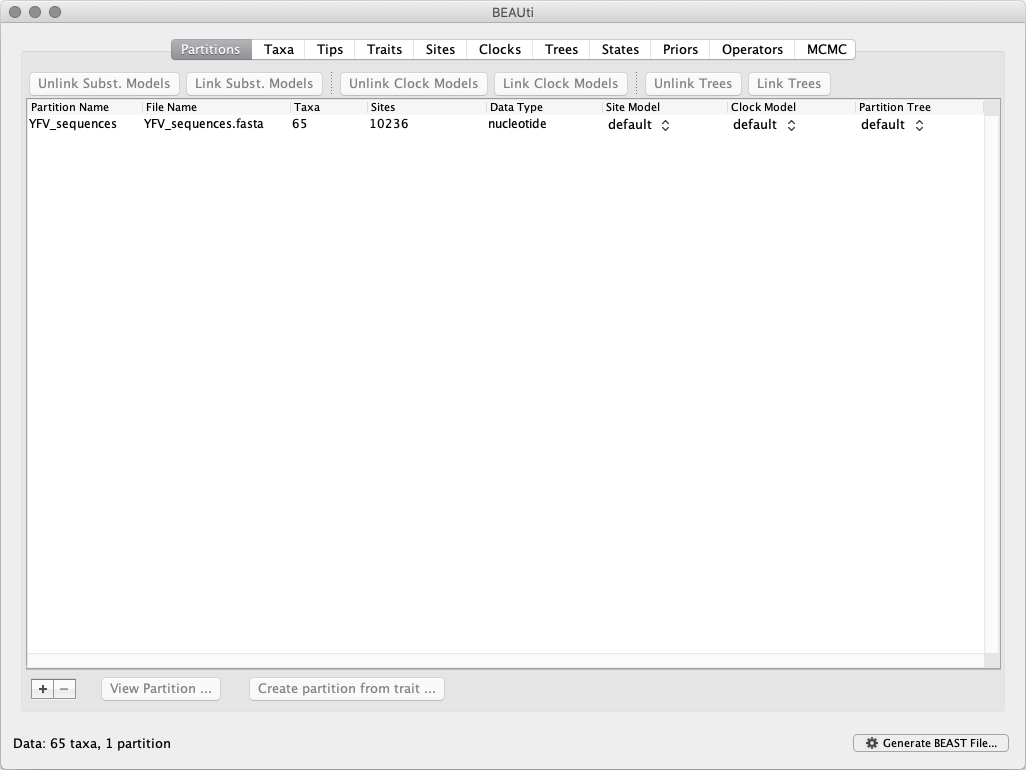
Specifying the sampling date information
By default all the taxa are assumed to have a date of zero (i.e. the sequences are assumed to be sampled at the same time). In this case, the YFV sequences have been sampled at various days during 2017. To set these dates switch to the Tips panel using the tabs at the top of the window.
Select the box labelled Use tip dates. The actual sampling time in years is encoded in the name of each taxon and we could simply edit the value in the Date column of the table to reflect these. However, if the taxa names contain the calibration information, then a convenient way to specify the dates of the sequences in BEAUti is to use the Parse Dates button at the top left of the Data panel. Clicking this will make a dialog box appear:
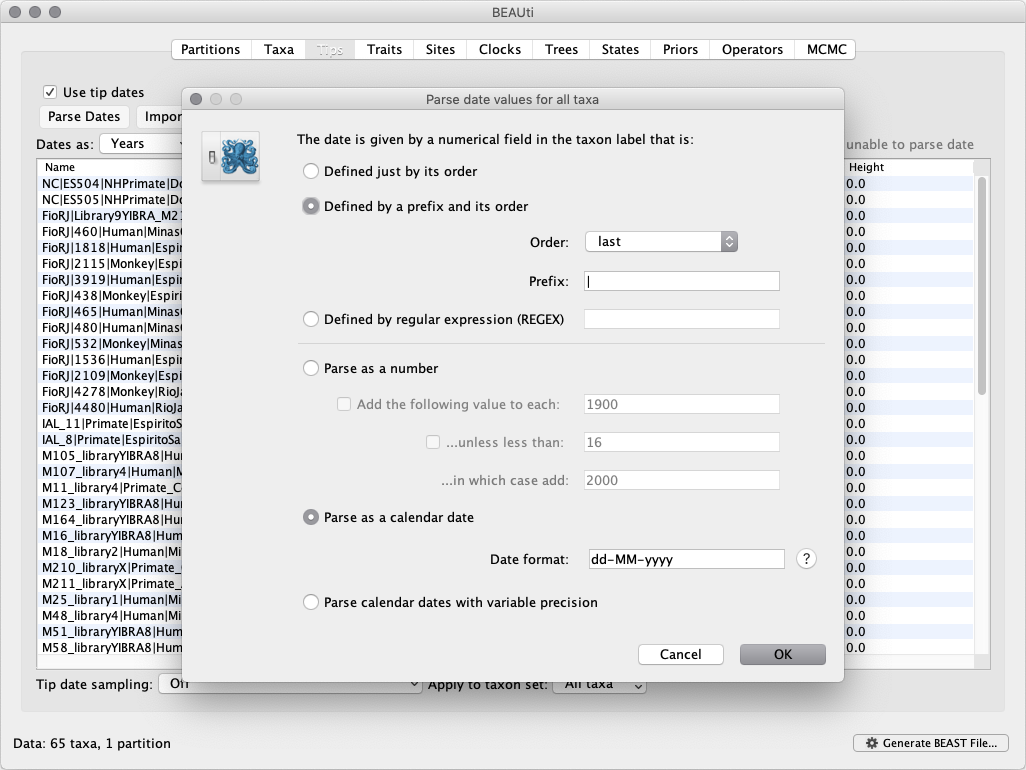
This operation attempts to guess what the dates are from information contained within the taxon names. It works by trying to find a numerical field within each name. If the taxon names contain more than one numerical field then you can specify how to find the one that corresponds to the date of sampling. You can (1) specify the order that the date field comes (e.g. first, last or various positions in between) or (2) specify a prefix (some characters that come immediately before the date field in each name) and the order of the field, or (3) define a regular expression (REGEX).
When parsing a number, you can ask BEAUti to add a fixed value to each guessed date. For example, the value 1900 can be added to turn the dates from 2 digit years to 4 digit years. Any dates in the taxon names given as 00 would thus become 1900. However, if these 00 or 01, etc. represent sequences sampled in 2000, 2001, etc., 2000 needs to be added to those. This can be achieved by selecting the unless less than: ... and ... in which case add: ... options adding for example 2000 to any date less than 10. These operations are not necessary in the present case because the dates are fully specified at the end of the sequence names. There is also an option to parse calendar dates and one for calendar dates with various precisions.
For the YFV sequences, dates values have to be parsed as calendar dates by specifying the format dd-MM-yyyy. For the other options, select Defined by a prefix and its order, select last from the drop-down menu for the order, set the Prefix to the symbol | and press OK. The dates will appear in the appropriate column of the main window.
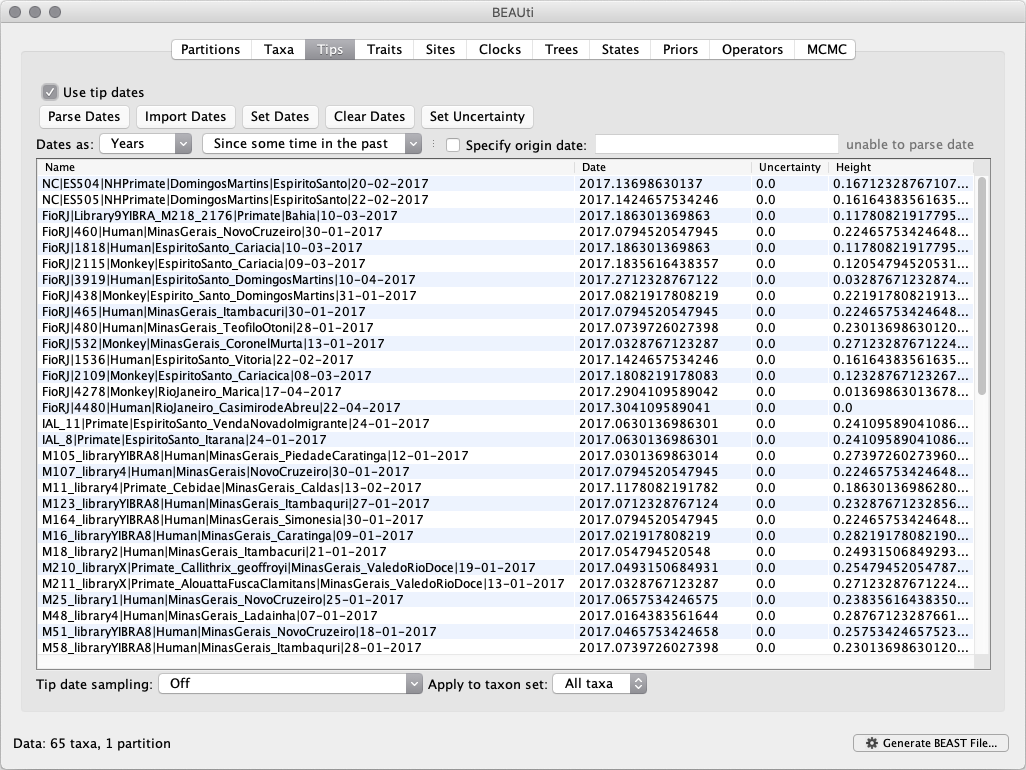
You can now check these and edit them manually as required. At the top of the window you can set the units that the dates are given in (years, months, days) and whether they are specified relative to a point in the past (as is the case for years such as 2017) or backwards in time from the present (as in the case of radiocarbon ages).
The Height column lists the ages of the tips relative to time 0 (in our case 2007.63). The Uncertainty column allows specifying with what precision the sampling time is known. This is not useful in our case because all sampling dates are known to the exact day, but BEAST allows to integrate over the uncertainty for less precise sampling date (e.g. when only the sampling month or year is known).
Specifying the trait information
The next thing to do is to click on the Traits tab at the top of the main window. A trait can be any characteristic that is inherent to the specific taxon, for example, geographical location or host species. This step will assign a latitude and longitude as bivariate geographical location to each taxa based on the trait specification for each sequence.
To associate the sequences with the traits, we need to import a new trait under the Traits tab (click Import Traits...). This will open a new window that allows importing a file with the traits. Browse to and Open the ‘YFV_coordinates.txt’ tab-delimited file, with the following content:
traits lat long
NC|ES504|NHPrimate|DomingosMartins|EspiritoSanto|20-02-2017 -20.360797 -40.65981
NC|ES505|NHPrimate|DomingosMartins|EspiritoSanto|22-02-2017 -20.360797 -40.65981
FioRJ|Library9YIBRA_M218_2176|Primate|Bahia|10-03-2017 -15.037801 -41.934681
FioRJ|460|Human|MinasGerais_NovoCruzeiro|30-01-2017 -17.432895 -41.998972
FioRJ|1818|Human|EspiritoSanto_Cariacia|10-03-2017 -20.284466 -40.431258
FioRJ|2115|Monkey|EspiritoSanto_Cariacia|09-03-2017 -20.284466 -40.431258
FioRJ|3919|Human|EspiritoSanto_DomingosMartins|10-04-2017 -20.360797 -40.65981
FioRJ|438|Monkey|Espirito_Santo_DomingosMartins|31-01-2017 -20.360797 -40.65981
FioRJ|465|Human|MinasGerais_Itambacuri|30-01-2017 -18.034092 -41.683337
FioRJ|480|Human|MinasGerais_TeofiloOtoni|28-01-2017 -17.860009 -41.509104
FioRJ|532|Monkey|MinasGerais_CoronelMurta|13-01-2017 -16.588013 -42.196772
FioRJ|1536|Human|EspiritoSanto_Vitoria|22-02-2017 -20.297618 -40.295777
...
MF170971|Monkey|MinasGerais_SaoRoqueDeMinas|NA|30-01-2017 -20.177911 -46.439305
After clicking OK, BEAuti asks to create a traits partition. Click Yes and select both the lat and long and provide a name for the triat parttition, e.g. ‘location’ and Click OK.
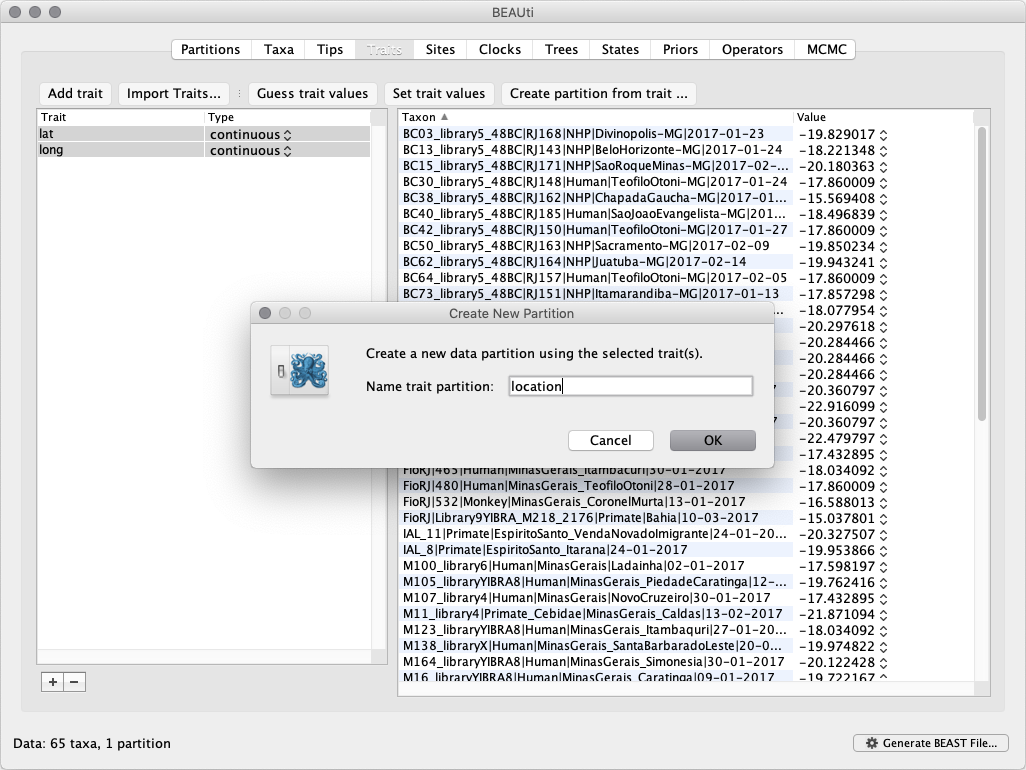
This new location partition with 2 Sites and with a continuous Data Type will be shown under the Partitions tab:
Setting the sequence and trait evolutionary models
The next thing to do is to click on the Sites tab at the top of the main window. This will reveal the evolutionary model settings for BEAST. Exactly which options appear depend on whether the data are nucleotides, amino acids or traits.
This tutorial assumes that you are familiar with the available evolutionary models, however there are a couple of points to note about selecting a model in BEAUti:
Selecting the Partition into codon positions option assumes that the data are aligned as codons. This option will then estimate a separate rate of substitution for each codon position, or for 1+2 versus 3, depending on the setting. Selecting the Unlink substitution model across codon positions will specify that BEAST should estimate a separate transition-transversion ratio or general time reversible rate matrix for each codon position. Selecting the Unlink rate heterogeneity model across codon positions will specify that BEAST should estimate a set of rate heterogeneity parameters (gamma shape parameter and/or proportion of invariant sites) for each codon position.
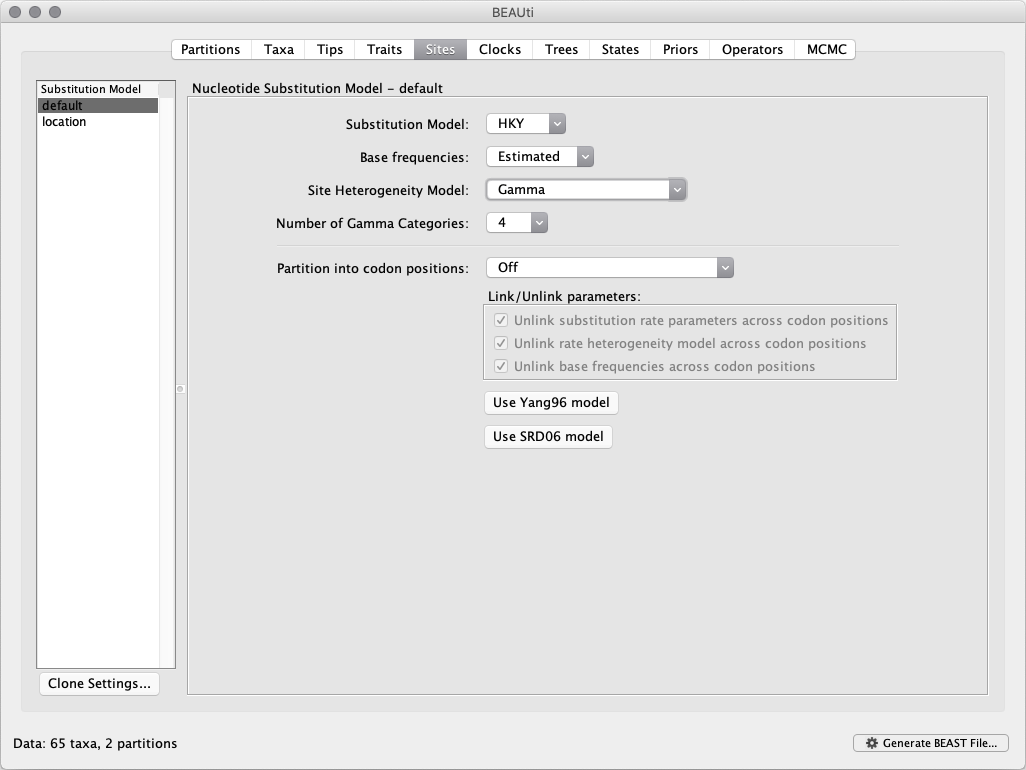
For the nucleotide substitution model in this tutorial, keep the default HKY substitution model, keep the base frequencies to be Estimated, specify the Site Heterogeneity Model to Gamma, and the keep the default Partition into codon positions to Off.
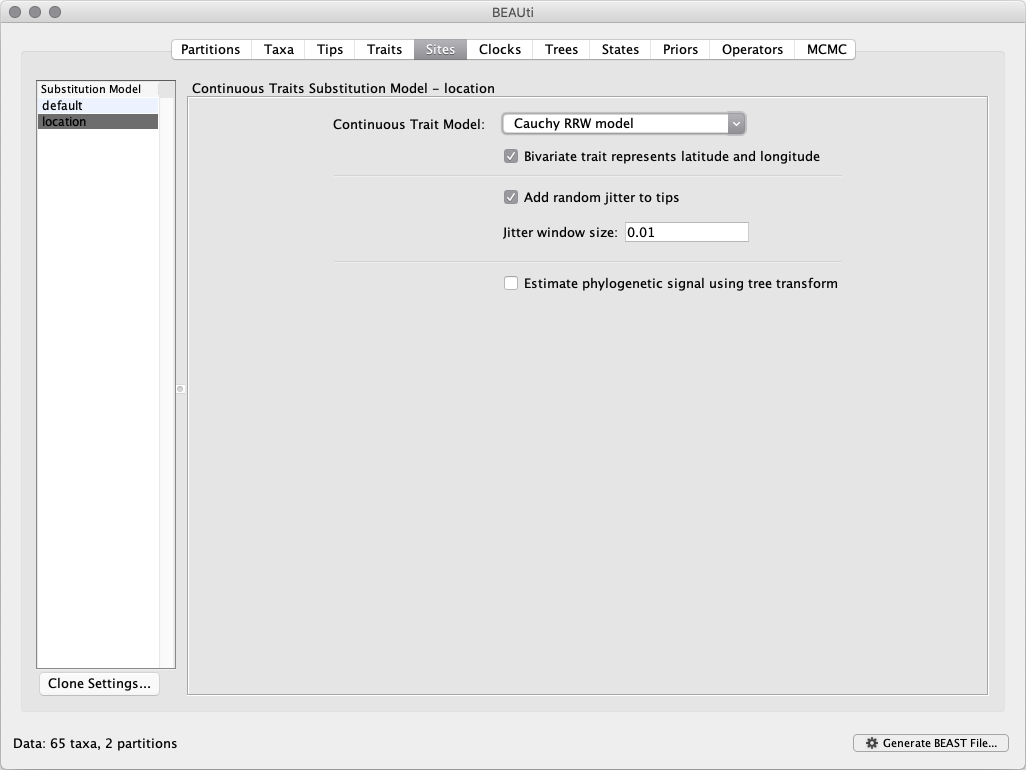
Next, click on location in the Substitution Model window and select the
Cauchy RRW model, and select Bivariate trait represents latitude and longitude. This option generates diffusion statistics that are specific for bivariate spatial traits (with latitude and longitude in that order). Also select the Add random jitter to tips, which adds noise drawn uniformly at random from a particular jitter window size to duplicated (location) traits. Set the Jitter window size to 0.01. The noise avoids a poor performance of Brownian diffusion models when not all sequences are associated with unique locations.
Setting the molecular clock model
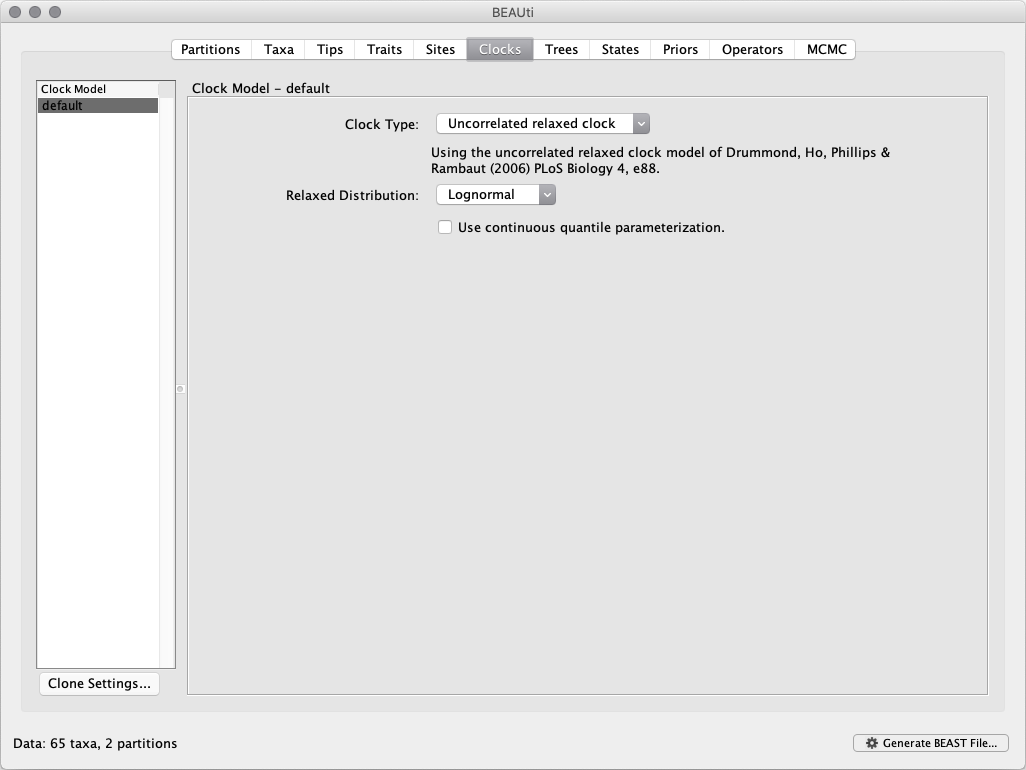
The molecular clock model options in the Clocks panel allows us to choose between a strict and a relaxed (uncorrelated lognormal or uncorrelated exponential) clock. We will perform our run using the lognormal relaxed molecular clock (uncorrelated) model:
Now move on to the Trees panel.
Setting the tree prior
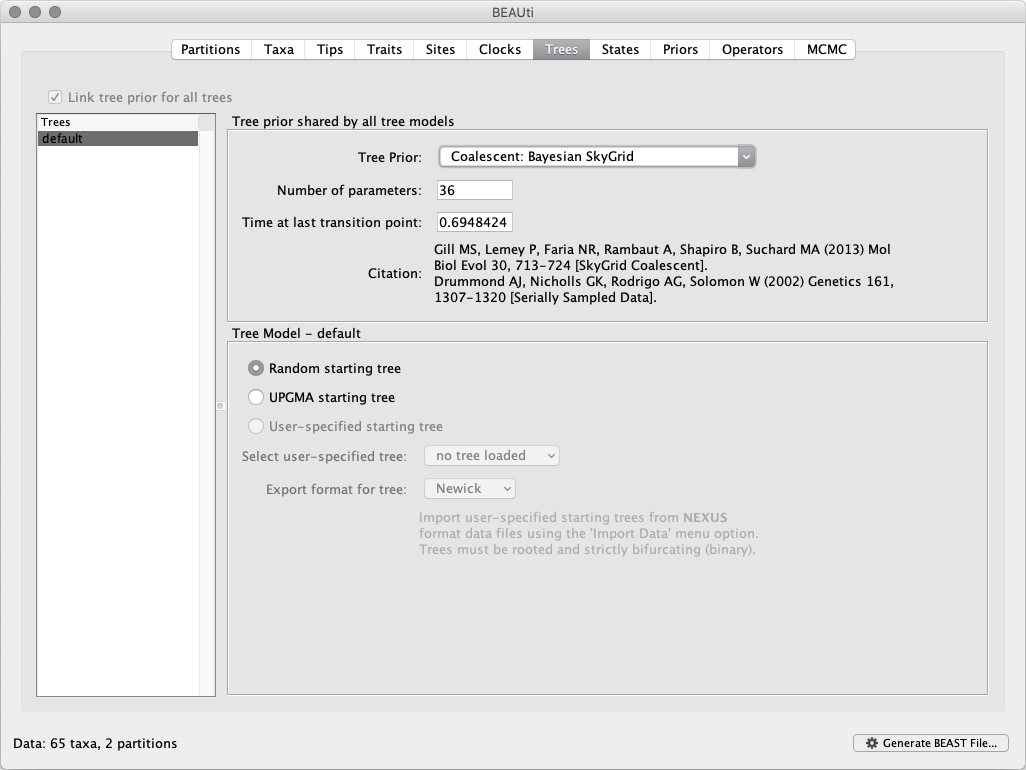
This panel contains settings about the tree. Firstly the starting tree is specified to be ‘randomly generated’. The other main setting here is to specify the Tree Prior, which describes how the population size is expected to change over time according to a coalescent model. The default tree prior is set to a constant size coalescent prior. Here, we will select a flexible skygrid coalescent model as demographic tree prior (Coalescent: Bayesian SkyGrid), with 36 grid points (Number of parameters) and Time at last transition point set to 0.6948424. By doing so, the grid points actually approximate the number of epidemiological weeks spanned by the duration of the phylogeny (Faria et al. 2018).
The ancestral states settings
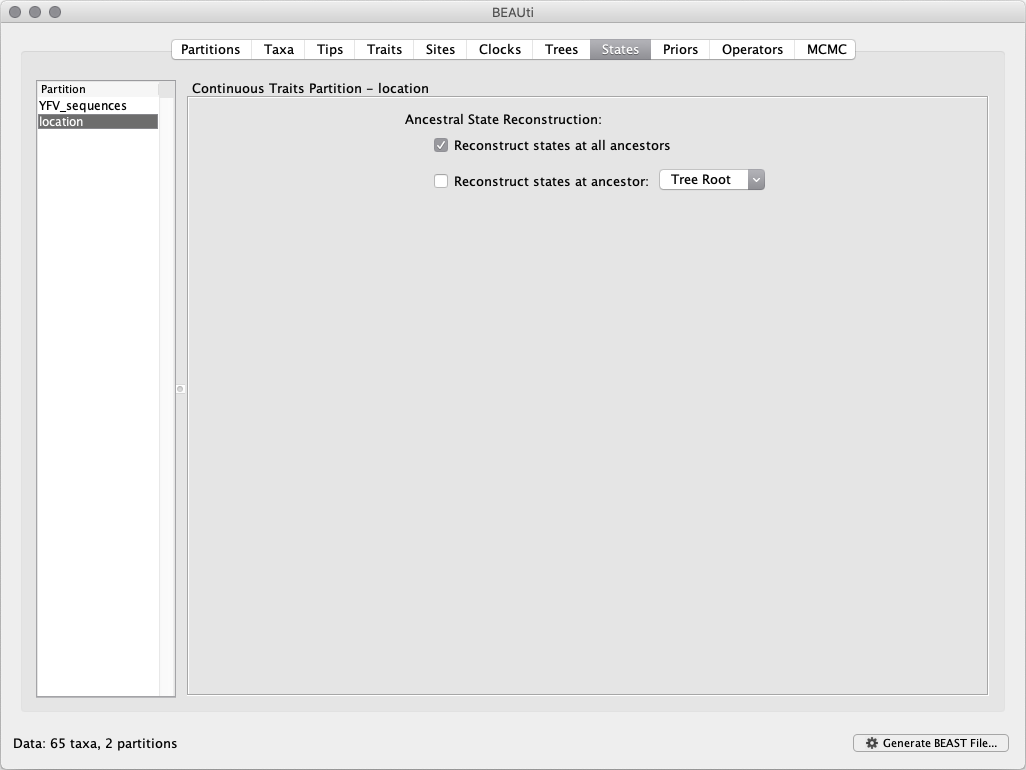
In the States panel, check that location partition is set to Reconstruct states at all ancestors (by default):
Setting up the priors
Review the prior settings under the Priors tab. This panel has a table showing every parameter of the currently selected model and what the prior distribution is for each.
Setting up the operators
Each parameter in the model has one or more ‘operators’ (these are variously called moves, proposals or transition kernels by other MCMC software packages such as MrBayes and LAMARC). The operators specify how the parameters change as the MCMC runs. The Operators tab in BEAUti has a table that lists the parameters, their operators and the tuning settings for these operators:
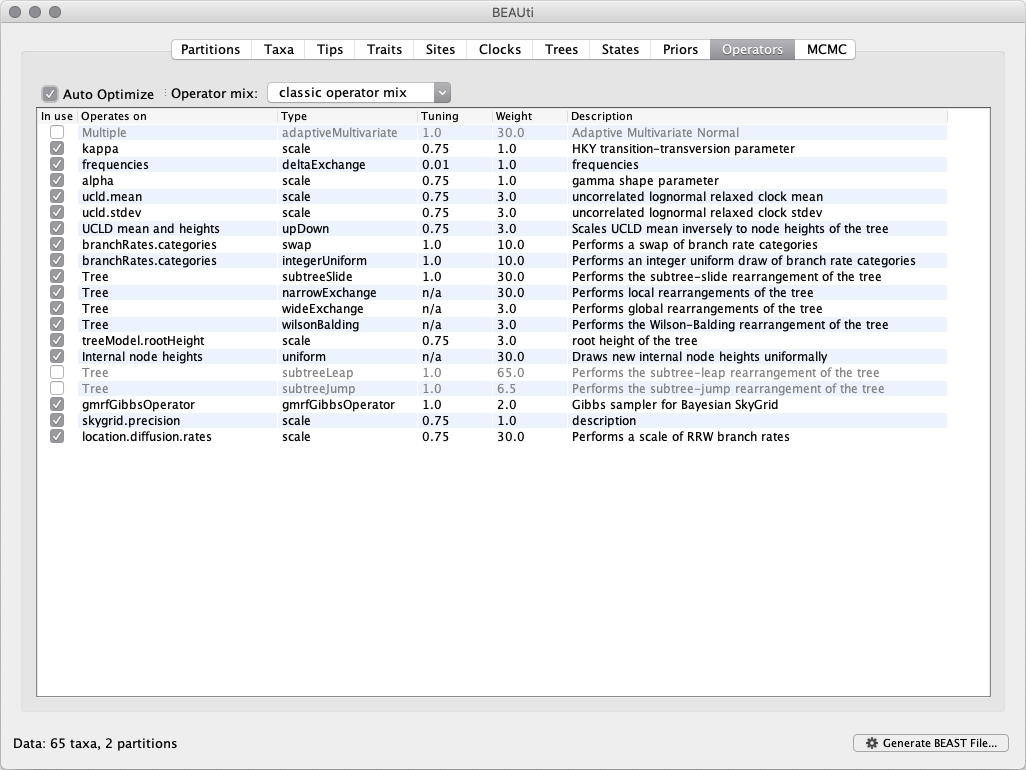
In the first column are the parameter names. These will be called things like ‘kappa’, which means the HKY model’s kappa parameter (the transition-transversion bias). The next column has the type of operators that are acting on each parameter. For example, the scale operator scales the parameter up or down by a proportion, the random walk operator adds or subtracts an amount to the parameter and the uniform operator simply picks a new value uniformly within a range. Some parameters relate to the tree or to the divergence times of the nodes of the tree and these have special operators. In this tutorial, we will use the classic operator mix, which consists of a set of tree transition kernels that propose changes to the tree. There is also an option to fix the tree topology as well as a new experimental mix, which is currently under development with the aim to improve mixing for large phylogenetic trees.
The next column, labelled Tuning, gives a tuning setting to the operator. Some operators do not have any tuning settings so have ‘n/a’ under this column. The tuning parameter will determine how large a move each operator will make, which will affect how often that change is accepted by the MCMC, and which will in turn affect the efficiency of the analysis. For most operators (like random walk and subtree slide operators) a larger tuning parameter means larger moves. However for the scale operator a tuning parameter value closer to 0.0 means bigger moves. At the top of the window is an option called Auto Optimize which, when selected, will automatically adjust the tuning setting as the MCMC runs to try to achieve maximum efficiency. At the end of the run, a table of the operators, their performance, and the final values of these tuning settings will be written to standard output.
The next column, labelled Weight, specifies how often each operator is applied relative to the others. Some parameters tend to be sampled very efficiently - an example is the kappa parameter - these parameters can have their operators down-weighted so that they are not changed as often. We will start by using the default settings for this analysis. As of BEAST v1.8.4, different options are available w.r.t. exploring tree space. In this tutorial, we will use the classic operator mix, which consists of a set of tree transition kernels that propose changes to the tree. There is also an option to fix the tree topology as well as a new experimental mix, which is currently under development with the aim to improve mixing for large phylogenetic trees. We can keep the default operator settings for the analysis in this tutorial.
Setting the MCMC options
The MCMC tab in BEAUti provides settings to control the MCMC chain. Firstly we have the Length of chain. This is the number of steps the MCMC will make in the chain before finishing. How long this should be depends on the size of the data set, the complexity of the model and the precision of the answer required. The default value of 10,000,000 is entirely arbitrary and should be adjusted according to the size of your data set. We will see later how the resulting log file can be analysed using Tracer in order to examine whether a particular chain length is adequate.
The next couple of options specify how often the current parameter values should be displayed on the screen and recorded in the log file. The screen output is simply for monitoring the program’s progress so can be set to any value (although if set too small, the sheer quantity of information being displayed on the screen will slow the program down). For the log file, the value should be set relative to the total length of the chain. Sampling too often will result in very large files with little extra benefit in terms of the precision of the estimates. Sample too infrequently and the log file will not contain much information about the distributions of the parameters. You probably want to aim to store no more than 10,000 samples.
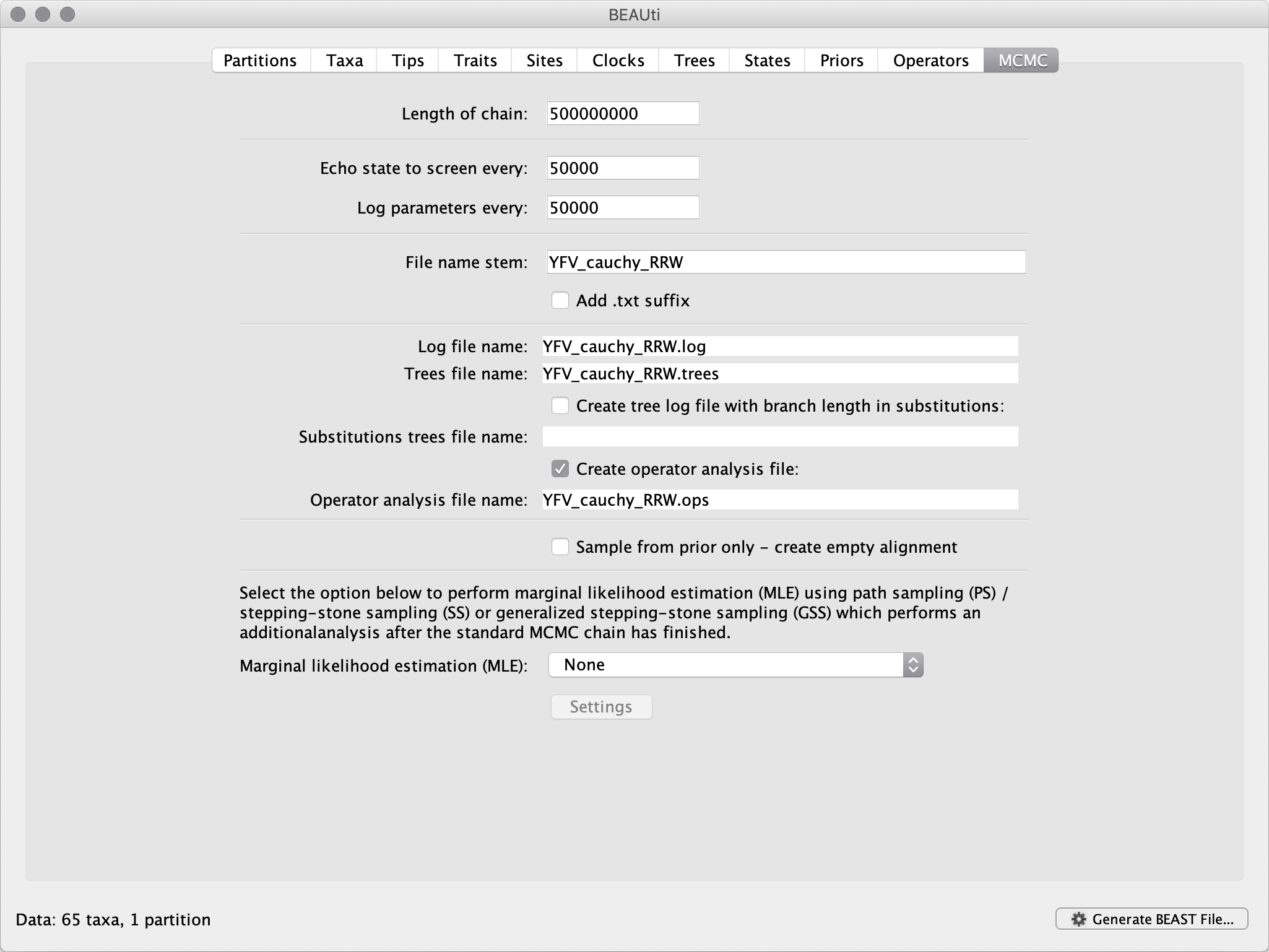
For this data set let’s set the chain length to 500,000,000 and the parameter logging to file as well the state echo to screen every 50,000 state.
The next option allows the user to set the File stem name; set this to ‘YFV_RRW_cauchy’ (‘YFV_RRW_cauchy’ for ‘relaxed random walk Cauchy diffusion model’). The next two options give the file names of the log files for the parameters and the trees. These will be set based on the file stem name. You can also log the operator analysis to a file. An option is also available to sample from the prior only, which can be useful to evaluate how divergent our posterior estimates are when information is drawn from the data. Here, we will not select this option, but analyse the actual data.
Finally, one can select an option to perform marginal likelihood estimation to assess model fit, which is not needed in this exercise. So, at this point we are ready to generate a BEAST XML file and to use this to run the Bayesian evolutionary analysis. To do this, either select the Generate BEAST File... option from the File menu or click the similarly labelled button at the bottom of the window. Continue and choose a name for the file (for example, ‘YFV_RRW_cauchy.xml’ by adding the xml extension to the file name stem) and save the file. For convenience, you can leave the BEAUti window open so that you can change the values and re-generate the BEAST file if necessary.
Running BEAST
Once the BEAST XML file has been created the analysis itself can be performed using BEAST. The exact instructions for running BEAST depends on the computer you are using, but in most cases a dialog box will appear in which you select the XML file:
Press the Choose File button and select the XML file you just created and press Run. When you have installed the BEAGLE library (https://github.com/beagle-dev/beagle-lib), you can use this in conjunction with BEAST to speed up the calculations. If not installed, unselect the use of the BEAGLE library. If the command line version of BEAST is being used then the name of the XML file is given after the name of the BEAST executable. The analysis will then be performed with detailed information about the progress of the run being written to the screen. When it has finished, the log file and the trees file will have been created in the same location as your XML file.
Analysing the BEAST output using Tracer
To analyse the results of running BEAST we are going to use the program Tracer. The exact instructions for running Tracer differs depending on which computer you are using. Double click on the Tracer icon; once running, Tracer will look similar irrespective of which computer system it is running on.
Select the Import Trace File... option from the File menu. If you have it available, select the log file that you created in the previous section (‘YFV_RRW_cauchy.log’). Alternative, drag and drop your log file into the Tracer window. The file will load and you will be presented with a window similar to the one below. Remember that MCMC is a stochastic algorithm so the actual numbers will not be exactly the same.
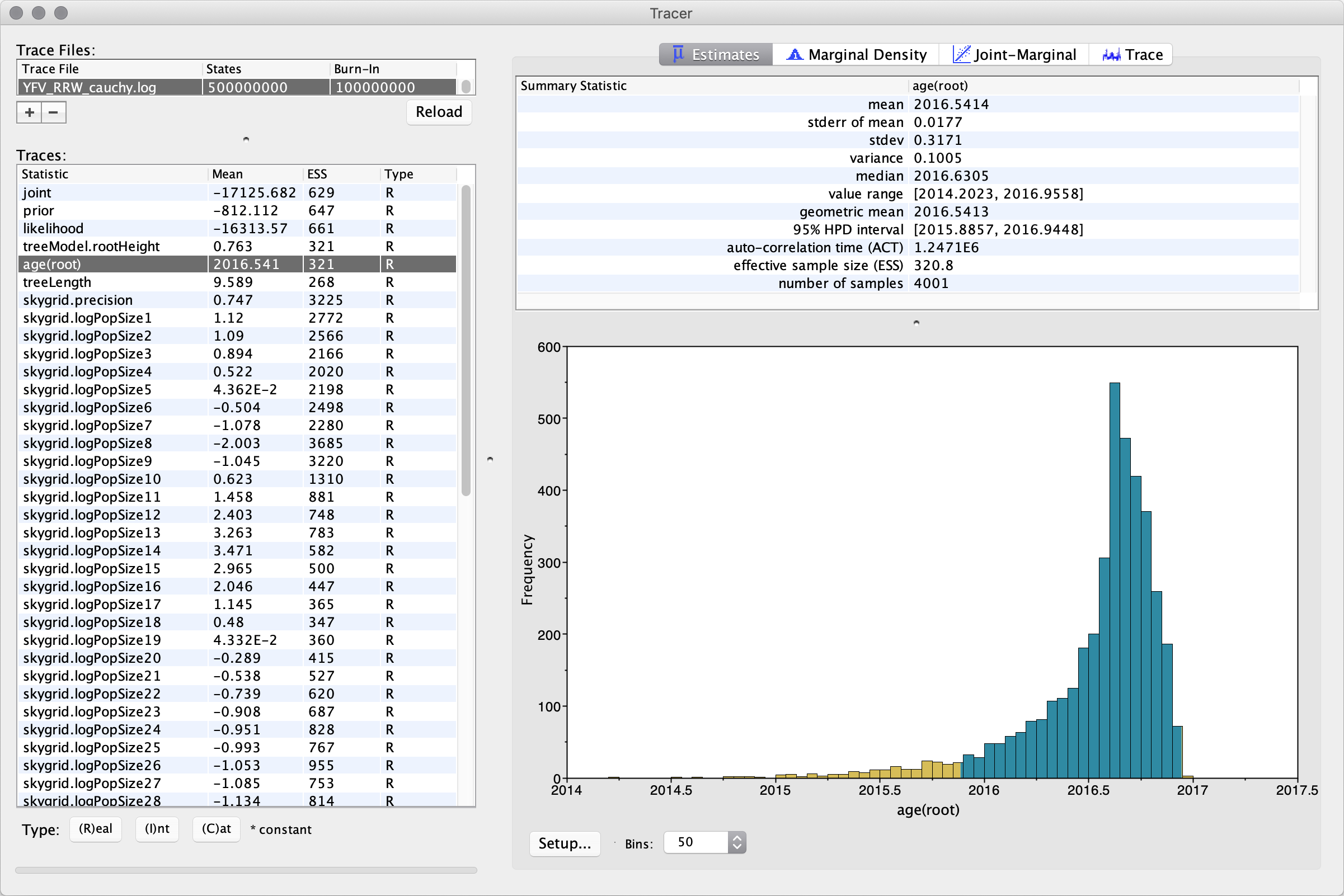
On the left hand side is the name of the log file loaded and the traces that it contains. Selecting a trace on the left brings up analyses for this trace on the right hand side depending on tab that is selected. When first opened, the age(root) trace is selected and various statistics of this trace are shown under the Estimates tab.
In the top right of the window is a table of calculated statistics for the selected trace. The statistics and their meaning are described in the table below.
-
mean: the mean value of the samples (excluding the burn-in) -
stdev: The standard error of the mean. This takes into account the effective sample size so a small ESS will give a large standard error -
median: the median value of the samples (excluding the burn-in) -
95% HPD lower: the lower bound of the highest posterior density (HPD) interval. The HPD is the shortest interval that contains 95% of the sampled values -
95% HPD upper: the upper bound of the highest posterior density (HPD) interval -
auto-correlation time (ACT): the average number of states in the MCMC chain that two samples have to be separated by for them to be uncorrelated (i.e. independent samples from the posterior). The ACT is estimated from the samples in the trace (excluding the burn-in) -
effective sample size (ESS): the effective sample size (ESS) is the number of independent samples that the trace is equivalent to. This is calculated as the chain length (excluding the burn-in) divided by the ACT
Note that when the effective sample sizes (ESSs) are small (ESSs less than 100 are highlighted in red by Tracer and values > 100 but < 200 are in yellow), this is not good. A low ESS means that your samples are equivalent to only few uncorrelated samples trace and this may not represent the posterior distribution well. A simple response to this kind of situation would be to run the chain for longer. Here, there are no obvious trends in the plot, which would suggest that the MCMC has not yet converged, and there are no large-scale fluctuations in the trace that would in turn suggest poor mixing.
Summarising the trees
We have seen how we can diagnose our MCMC run using Tracer and produce estimates of the marginal posterior distributions of parameters of our model. However, BEAST also samples trees (either phylogenies or genealogies) at the same time as the other parameters of the model. These are written to a separate file called the ‘trees’ file. This file is a standard NEXUS format file. As such it can easily be loaded into other software in order to examine the trees it contains. One possibility is to load the trees into a program such as PAUP* and construct a consensus tree in a similar manner to summarising a set of bootstrap trees. In this case, the support values reported for the resolved nodes in the consensus tree will be the posterior probability of those clades.
In this tutorial, however, we are going to use a tool that is provided as part of the BEAST package to summarise the information contained within our sampled trees. The tool is called TreeAnnotator and once running, you will be presented with a window like the one below.
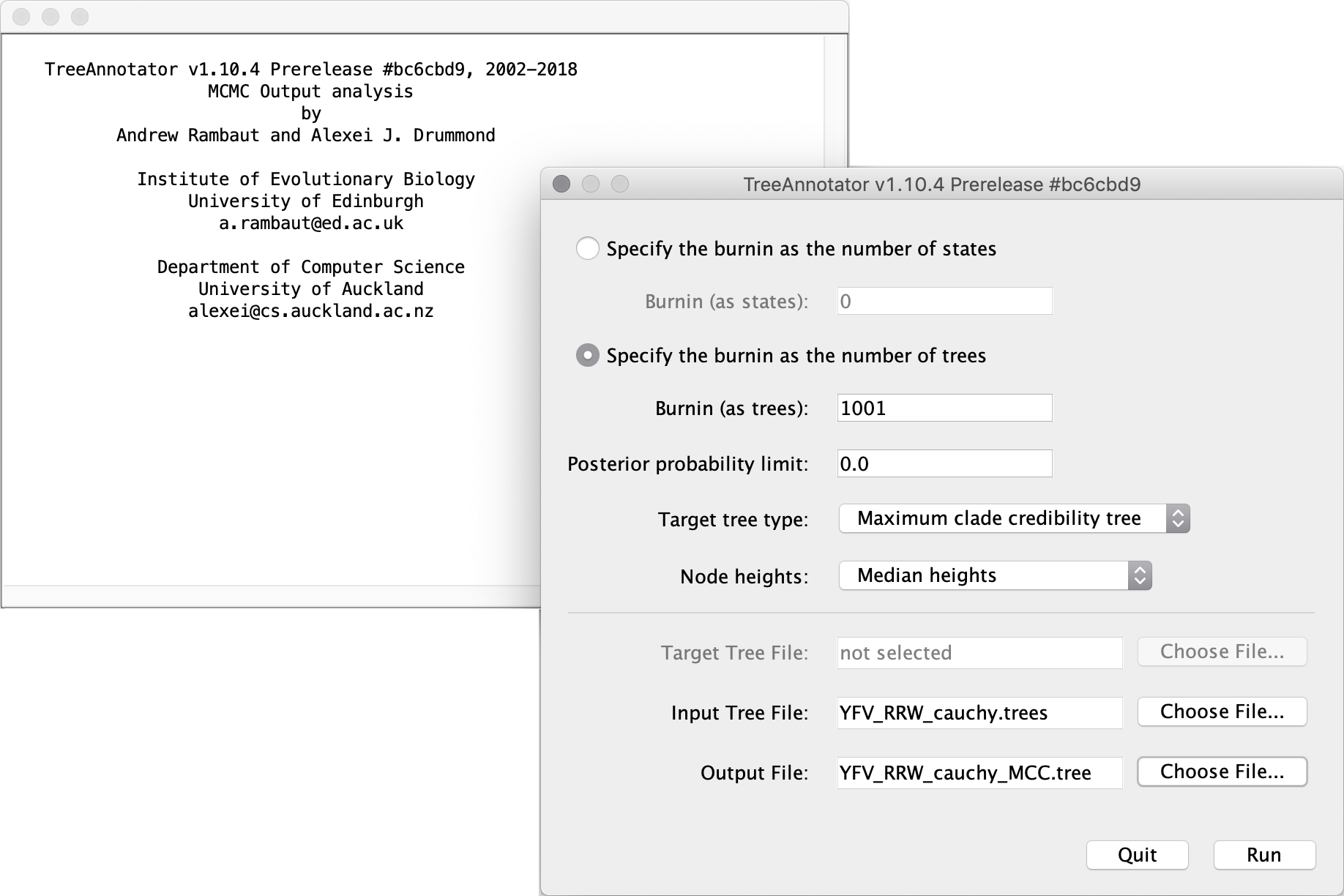
TreeAnnotator takes a single ‘target’ tree and annotates it with the summarised information from the entire sample of trees. The summarised information includes the average node ages (along with the HPD intervals), the posterior support and the average rate of evolution on each branch (for relaxed clock models where this can vary). The program calculates these values for each node or clade observed in the specified ‘target’ tree. It has the following options:
-
Burnin: the number of steps in the MCMC chain,Burnin (as states), or the number of trees,Burnin (as trees), that should be excluded from the summarisation -
Posterior probability limit: the minimum posterior probability for a node in order for TreeAnnotator to store the annotated information. The default is 0.0 so every node, no matter what its support, will have information summarised. Make sure this value remains 0.0 as every node will require location annotation for further visualisation -
Target tree type: this has three optionsMaximum clade credibility treeorUser target treeFor the latter option, a NEXUS tree file can be specified as the Target Tree File, below. Select the first option, TreeAnnotator will examine every tree in the Input Tree File and select the tree that has the highest product of the posterior probabilities of all its nodes -
Node heights: an option specifies what node heights (times) should be used for the output tree. If theKeep target heightsis selected, then the node heights will be the same as the target tree. Node heights can also be summarised as a Mean or a Median over the sample of trees. Sometimes a mean or median height for a node may actually be higher than the mean or median height of its parental node (because particular ancestral-descendent relationships in the MCC tree may still be different compared to a large number of other tree sampled). This will result in artifactual negative branch lengths, but can be avoided by theCommon Ancestor heightsoption. Let’s use the default Median heights for our summary tree -
Target Tree File: if theUser target treeoption is selected then you can useChoose File...to select a NEXUS file containing the target tree
Input Tree File - Use the Choose File... button to select an input trees file. This will be the trees file produced by BEAST
Output File- Select a name for the output tree file (e.g. ‘YFV_RRW_cauchy_MCC.tree’)
Once you have selected all the options, above, press the Run button. TreeAnnotator will analyse the input tree file and write the summary tree to the file you specified. This tree is in standard NEXUS tree file format so may be loaded into any tree drawing package that supports this. However, it also contains additional information that can only be displayed using the FigTree program.
Viewing the annotated tree
Run FigTree and select the Open... command from the File menu. Select the tree file you created using TreeAnnotator in the previous section. The tree will be displayed in the FigTree window. On the left hand side of the window are the options and settings, which control how the tree is displayed.
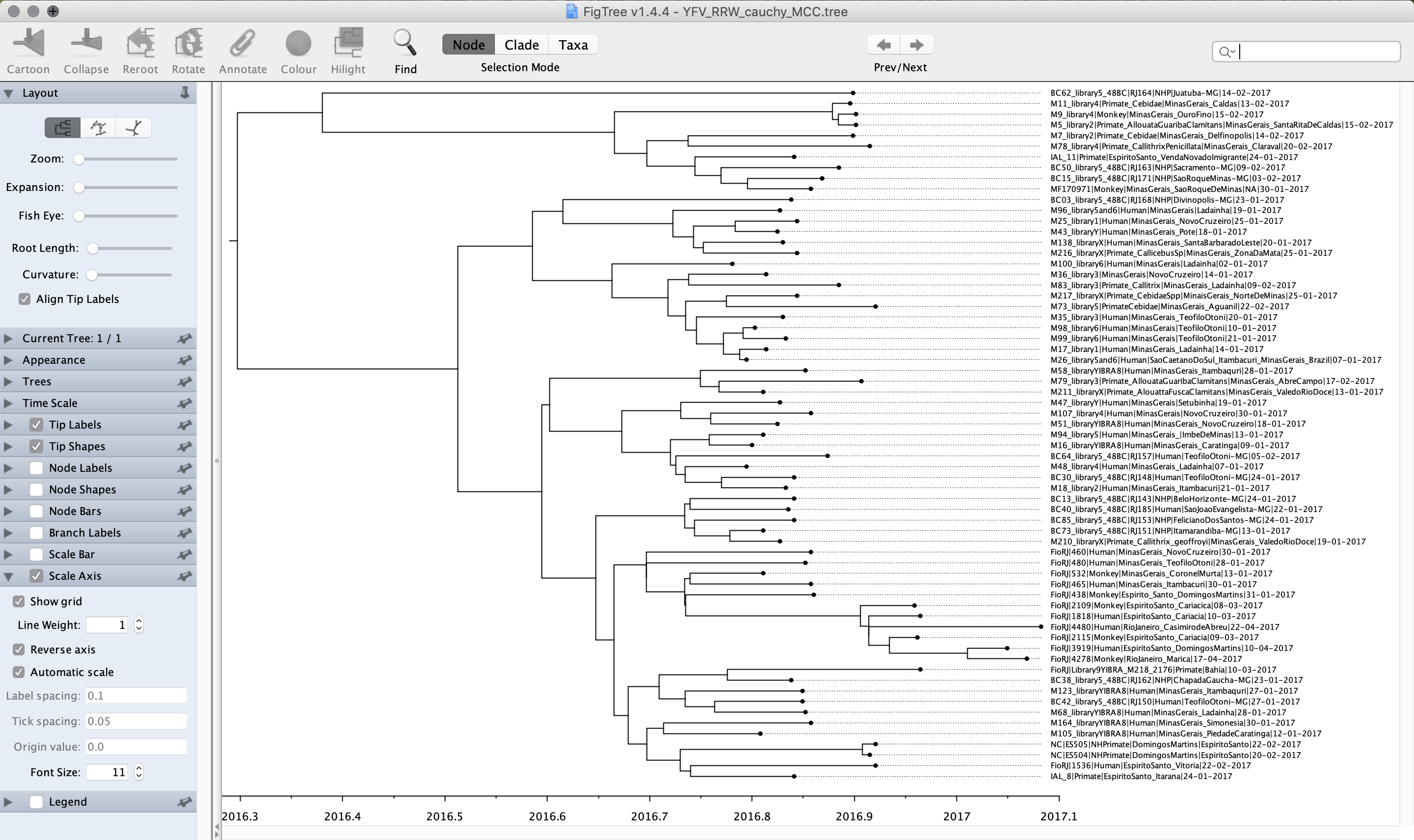
In the Layout panel select the check-box Align Tip Labels to increase clarity. We can also plot a time scale axis for this evolutionary history (select Scale Axis and deselect Scale Bar).
Visualizing Bayesian phylogeographic reconstructions using SPREAD4
SPREAD4, i.e. Spatial Phylogenetic Reconstruction of EvolutionAry Dynamics version 4, is a software to visualize the output from Bayesian phylogeographic analysis and constitutes a user-friendly application to analyze and visualize reconstructions resulting from Bayesian inference of sequence and trait evolutionary processes. SPREAD4 allows to visualise spatial reconstructions on custom maps and is run entirely online in browsers such as Firefox, Safari and Chrome.
Brief instructions can be found in the quick how-to summary below. Compare the animated version for both the homogeneous and RRW model: do you notice any difference?
A quick how-to summary
Run BEAUti
-
Load a NEXUS format alignment by selecting the
Import Data...option from the File menu. Select the file called ‘YFV_sequences.fasta’ -
In the
Tipstab, select theUse tip datesoption and use theParse Datesbutton. In the new window, select the optionDefined by a prefix and its order, and thelastfor the order. Finally, set thePrefixto the symbol|and pressOK. The dates will appear in the appropriate column of the main window -
In the
Traitstab, clickImport trait. Browse to and load the ‘YFV_coordinates.txt’ tab-delimited file -
After loading the file, select both the
LatandLongtraits in the panel on the left and selectCreate partition from trait...Enter the name ‘location’ for this partition -
In the
Sitestab, keep the default HKY substitution model, keep the base frequencies to be Estimated, and theSite Heterogeneity ModeltoGamma -
Click on location in the
Substitution modelwindow on the left and keep the defaultHomogenous Brownian modeland selectBivariate trait represents latitude and longitudeand aJitterset to ‘0.01’ -
In the
Clockstab, select the Lognormal relaxed molecular clock (Uncorrelated) model -
In the
Treestab, select the skygrid model as the demographic tree prior (Coalescent: Bayesian SkyGrid), with 36 grid points (number of parameters) andA time at last transition pointset to0.6948424(to define grid points approximating the number of epidemiological weeks spanned by the duration of the phylogeny) -
In the
MCMCtab, set the chain length to 500,000,000 and the sampling frequencies to 50,000. Set the File name stem to ‘YFV_RRW_cauchy’ and generate the beast file (‘YFV_RRW_cauchy.xml’)
Run BEAST and monitor output using Tracer
Once the BEAST XML file has been created, the analysis itself can be performed using BEAST. A common practice is to regularly monitor how much the analysis has progressed by loading the .log file(s) into Tracer and checking the ESS values of the (continuous) parameters. When running BEAST for a short amount of time, you will notice that - with the exception of very small examples - most ESS values remain far below the cut-off of 200 and will hence be colored in red in Tracer.
Analyse the output file for the longer runs
To get a good sense of what a completed analysis looks like in Tracer, load the provided output files (into Tracer).
Summarise the trees for the longer run using TreeAnnotator and visualise the result in FigTree
Construct a maximum clade credibility (MCC) tree in TreeAnnotator (check the required burn-in first) and load the result into FigTree. Set the time axis and play around with the node order, node labels and branch labels to obtain a visualisation that best showcases the important aspects of the MCC tree.
Run SPREAD 4
We will first summarise an MCC tree and then summarise the information in the entire tree distribution.
-
Select the
Continuous MCC treetab, load your MCC tree file -
Select ‘location2’ (longitude) in the
Select x coordinatebox and ‘location1’ (latitude) in theSelect y coordinatebox -
Set the
most recent sampling dateto 2017-04-22, and clickStart analysis

- There is an option to load a custom map of the United States in GeoJSON format. Such a map is provided amongst the data files –‘Brazil_states.geojson’. However, a default map is provided to visualize the results. Click
Copyand open the link in a new browser tab/window.
This visualises the MCC tree an the uncertainty for its node locations:

Conclusion and Resources
This tutorial only scratches the surface of the analyses that are possible to undertake using BEAST. It has hopefully provided a relatively gentle introduction to the fundamental steps that will be common to all BEAST analyses and provide a basis for more challenging investigations. BEAST is an ongoing development project with new models and techniques being added on a regular basis. The BEAST website provides details of the mailing list that is used to announce new features and to discuss the use of the package. The website also contains a list of tutorials and recipes to answer particular evolutionary questions using BEAST as well as a description of the XML input format, common questions and error messages.