Identifying convergence problems using Tracer
In a previous tutorial, analysing the output of a standard BEAST analysis using Tracer 1.7 was discussed. Here, this tutorial is extended towards analysing multiple independent runs (using the same combination of models), using different starting seeds (which leads to different random draws for all parameters involved to act as the initial estimates), in Tracer 1.7.
Data set information
Here, we take a look at an analysis of a (n unpublished) simulated data set based on a hepatitis C virus (HCV) data set, which consists of 378 taxa sampled between 1965 and July 2012. Two independent BEAST analyses (i.e. analysing the same simulated data set but with different starting seeds) were performed on this simulated data set, each comprising 90 million iterations, and should hence converge to the same posterior distribution. Comparison of marginal distributions and/or traces from multiple replicate chains can assist in diagnosing problems with phylogenetic MCMC analyses, given that well-behaved replicate chains will lead to similar posterior distributions.
Loading output files into Tracer
We start by loading the output .log files of both replicates of the same BEAST XML into Tracer 1.7. To load the log file(s), select the Open option from the File menu or drag and drop the log file into the Tracer window. The files will load and you will be presented with a window similar to the one below.
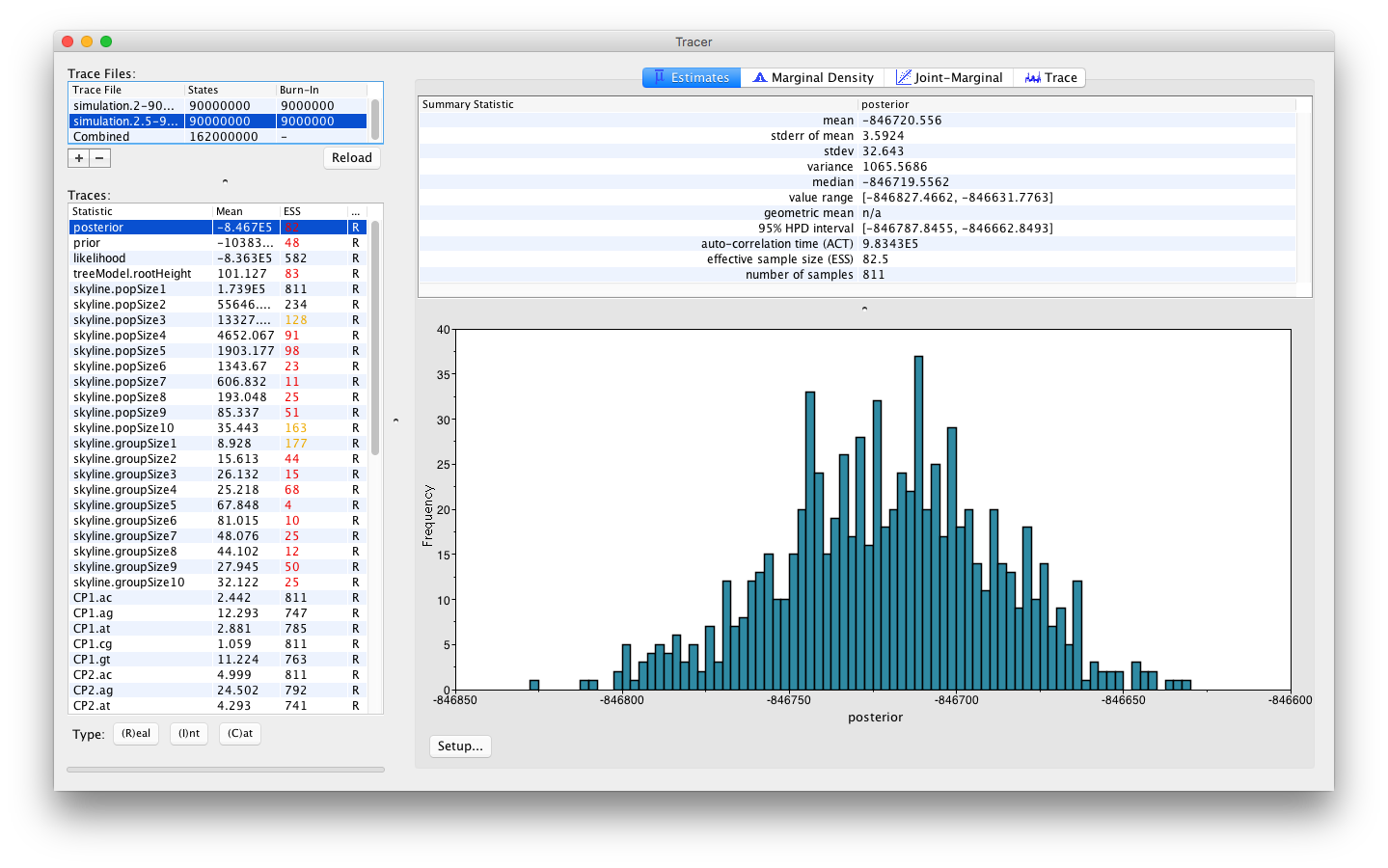
As with loading a single log file, the name of the log file loaded and the traces that it contains can be seen on the left hand side. When the different files loaded contain the same set of logged parameters, then a Combined trace will automatically appear (with a number of iterations equal to the sum of the two traces minus their burn-in length).
Exploring combined traces
Selecting the Combined trace allows to explore a concatenation of the log files. For the example we’re exploring here, selecting the Combined trace will yield the following visualisation:
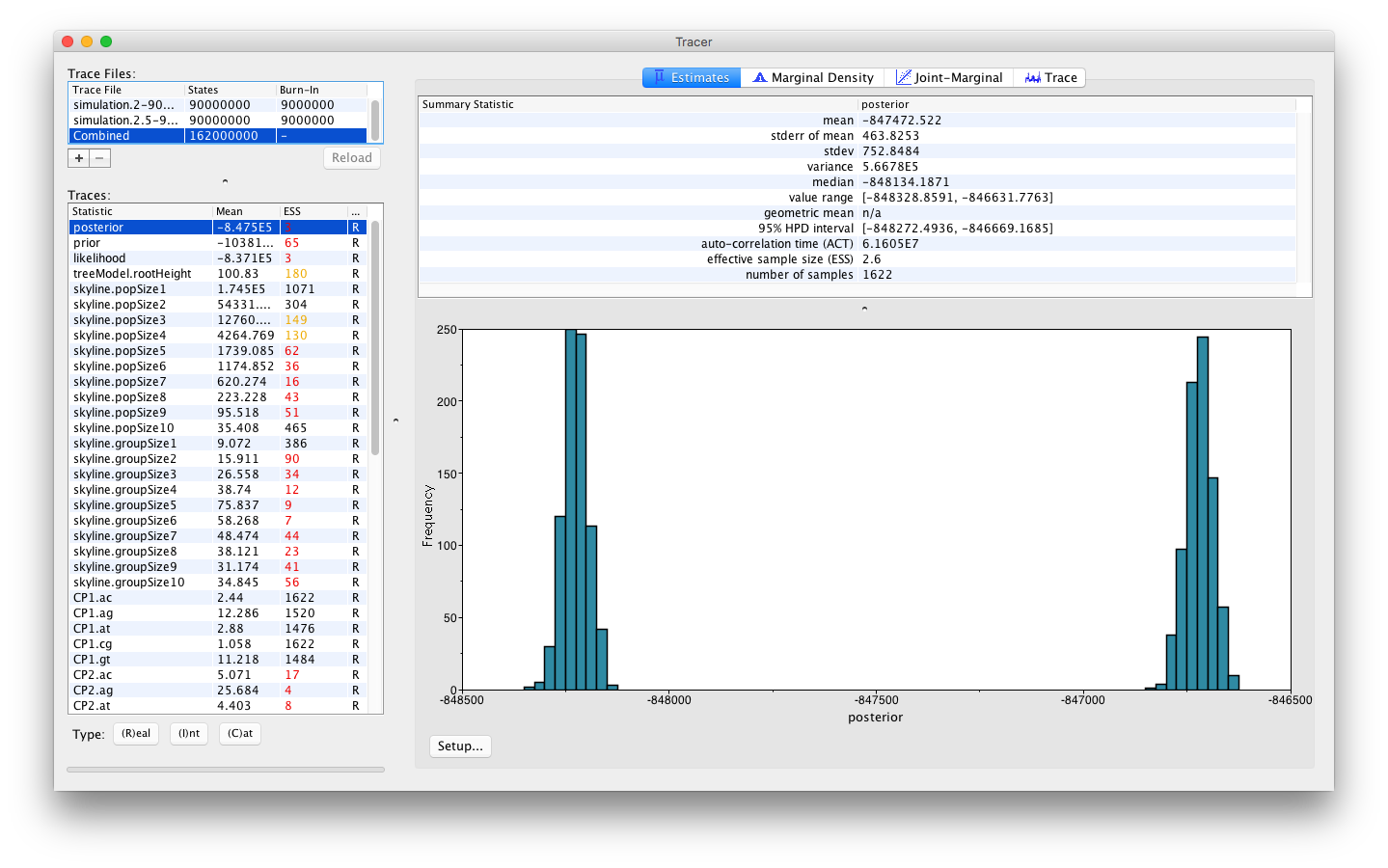
The histogram shown indicates a clear problem with the analysis, as the distribution of the joint density (or posterior) is bimodal. To obtain a better understanding of what’s going on, we can select the Trace panel which will show the following combined trace:
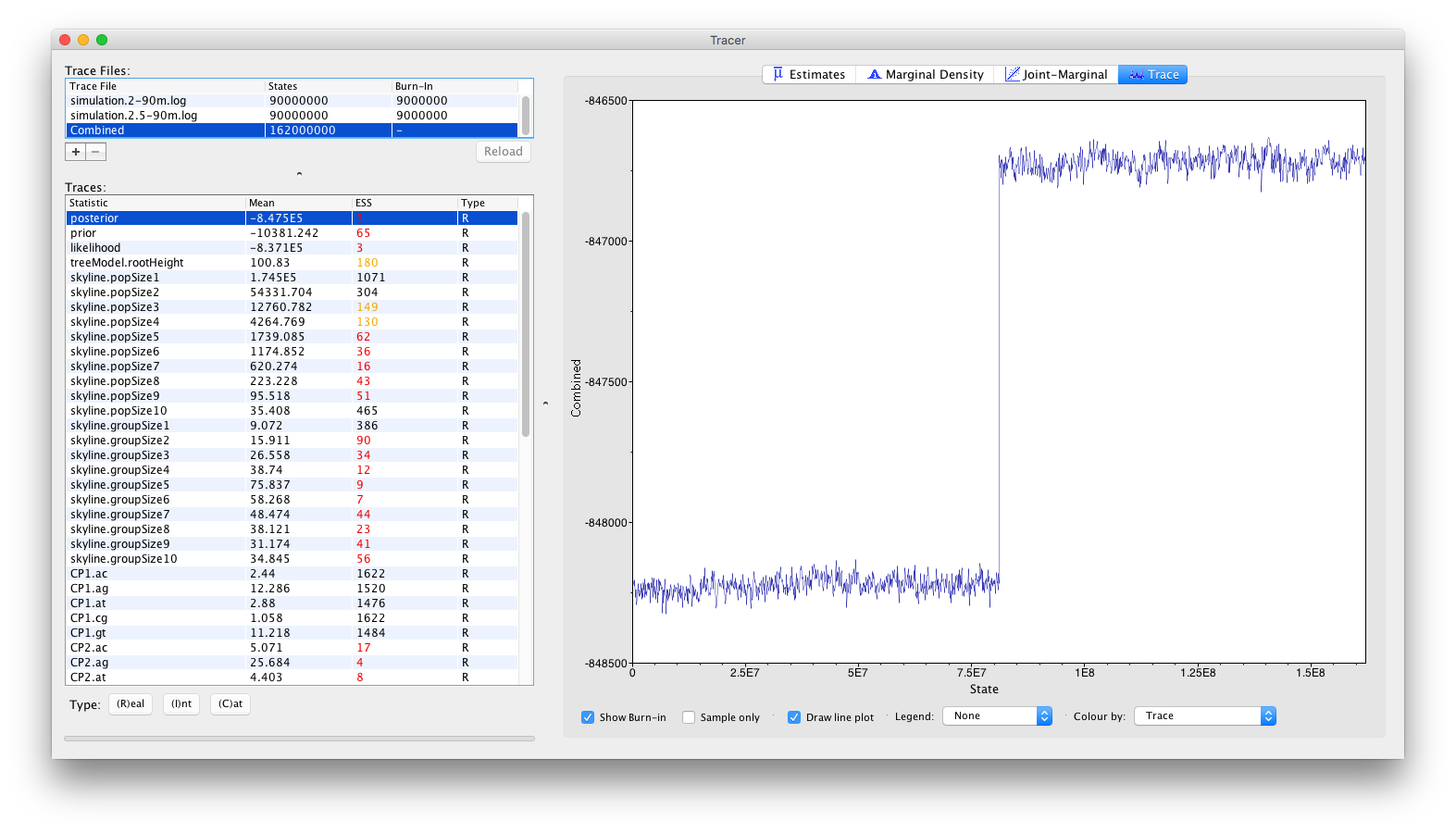
One of the joint density (or posterior) traces has converged around -848250, while the other has converged around -846750. This clearly showcases the need to perform multiple independent replicates of a Bayesian analysis, in order to be able to spot these types of problems.
Comparing individual traces
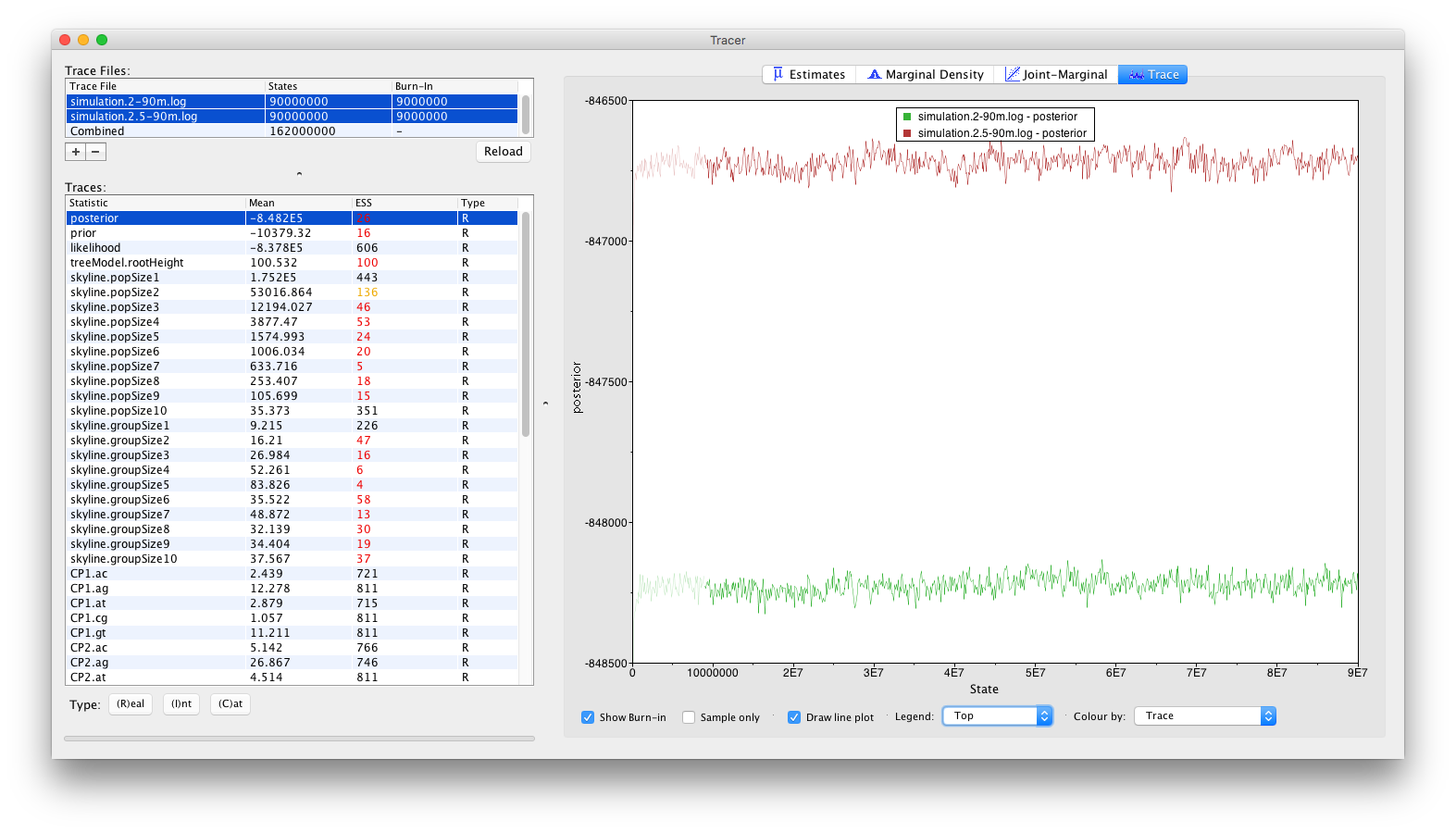
A more visually appealing approach to diagnose convergence problems is to not inspect the Combined trace, but rather inspecting the individual traces simultaneously. To this end, select the two trace files rather than the combined trace, which will generate the visualisation below. Select ‘Top’ in the Legend combo box at the bottom of the window to identify which trace belongs to which log file.
Alternative visualisations can be obtained by inspecting the Marginal Density panel:
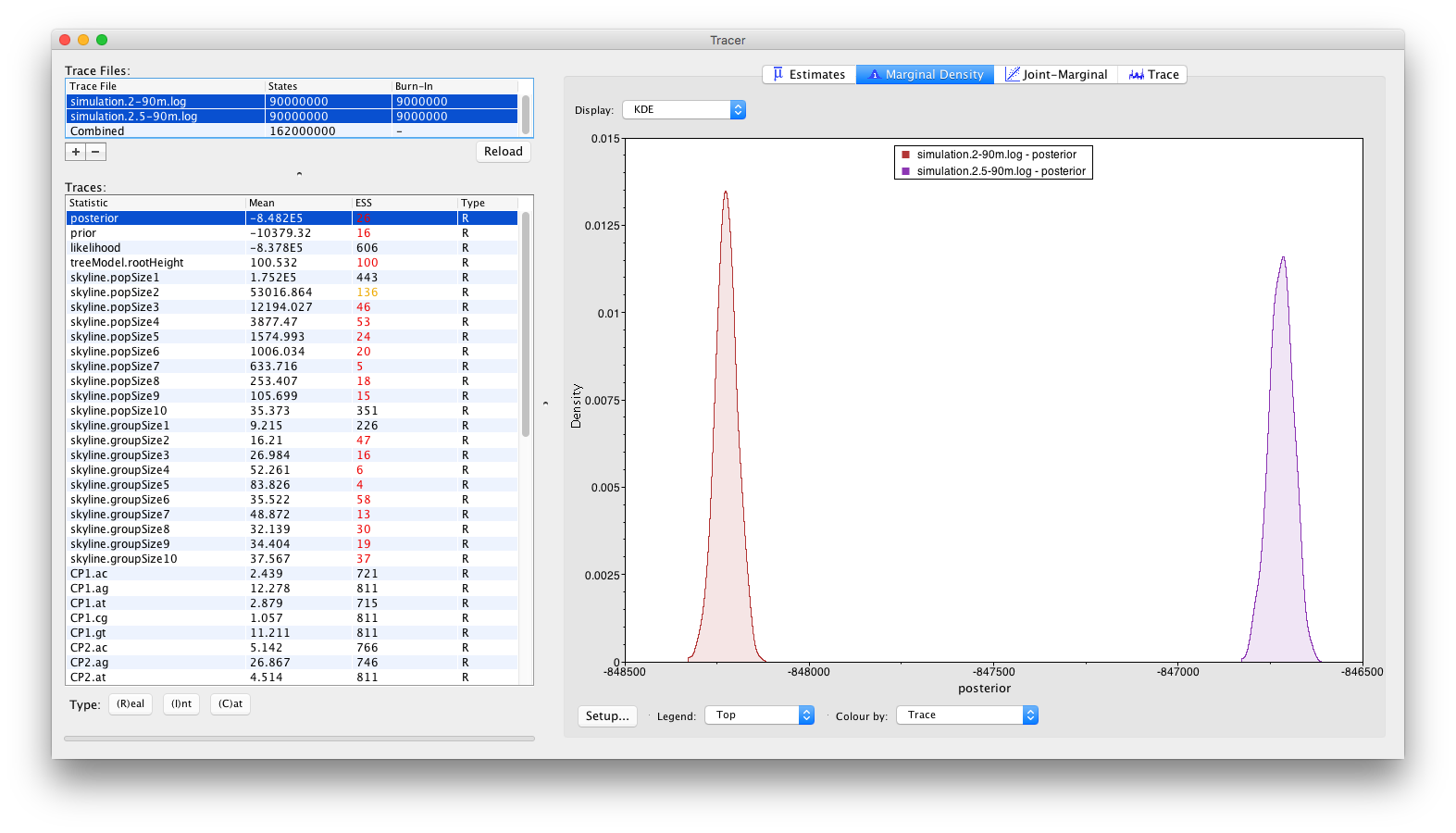
and by inspecting the Estimates panel, which allows to visualise/summarize the parameter values as either classical box plots:
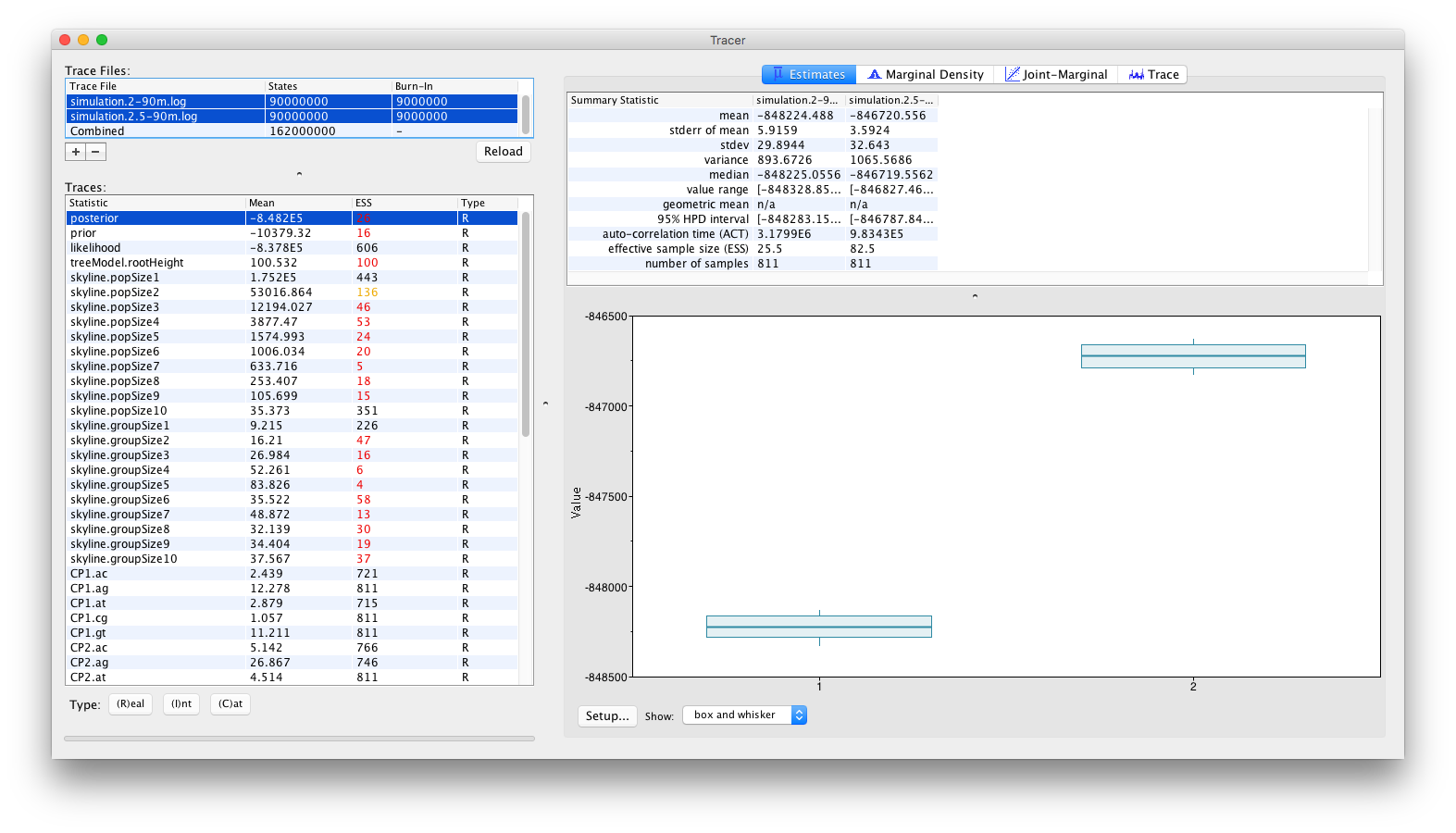
or as violin plots:
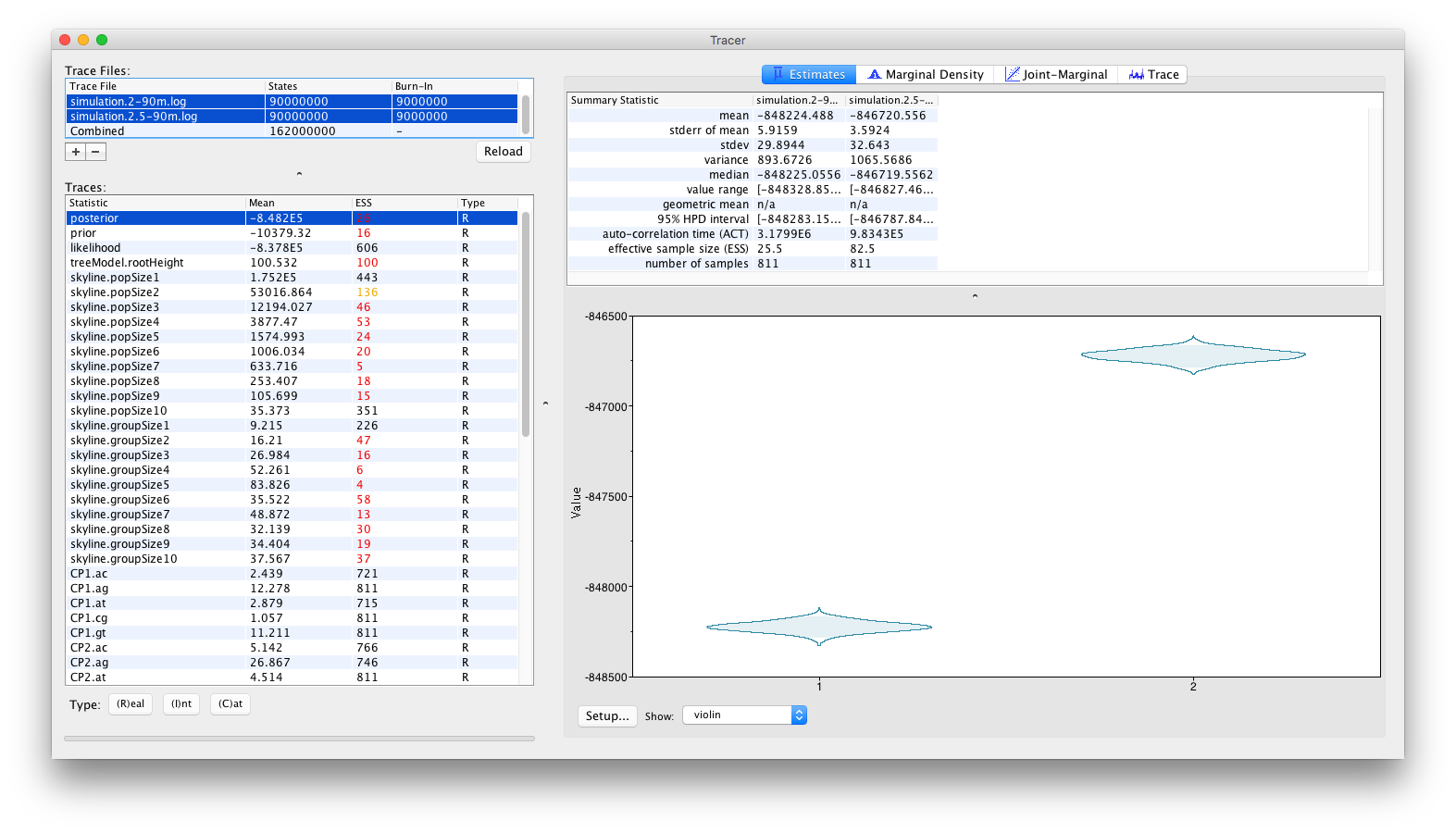
From these visualisations, it can clearly be concluded that after running for 90 million iterations, there is no sign of the replicate analyses to converge to the same joint density (or posterior). Obviously, with the current state of the analyses, no conclusions can be drawn as to which of these log files (if either) contain samples from the correct joint density. These types of convergence problems can be solved by a number of different approaches. The most obvious proposed solution would be to run the analyses for longer, in the hope that both replicate analyses convergence to the same joint density. Alternatively, it can be argued that both analyses are stuck in different (possibly local) optima and that the transition kernels need to be adjusted so that the Markov chains can escape these local optima. For example, a transition kernel that occasionally proposes a large jump across parameter space for one or more parameters may allow these analyses to converge towards the same joint density. In order to identify for which parameters this would be useful, careful inspection of the different parameter traces may be useful (to check which parameters clearly differ in their posterior estimates or if one or more parameters differ between replicate analyses).
Identifying convergence problems using TreeTracer
Data set information
Loading output files into TreeTracer
We start by loading the output .trees files of both replicates of the same BEAST XML into TreeTracer.
Citing Tracer and TreeTracer
The recommended citation for Tracer is:
Rambaut A, Drummond AJ, Xie D, Baele G and Suchard MA (2018) Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Systematic Biology. syy032. doi:10.1093/sysbio/syy032
The recommended citation for TreeTracer is:
References
Nylander, J. A. A., Wilgenbusch, J. C., Warren D. L., Swofford, D. L. (2007) AWTY (Are We There Yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 24(4):581-583.
Warren, D. L, Geneva, A. J., Lanfear, R. (2017) RWTY (R We There Yet): an R package for examining convergence of Bayesian phylogenetic analyses. Mol. Biol. Evol. 34(4):1016-1020.